
Effects of Heat Treatment on Phase Formation in Cytocompatible Sulphate-Containing Tricalcium Phosphate Materials
 , , ,
, , ,
Abstract
:
1. Introduction
2. Materials and Methods
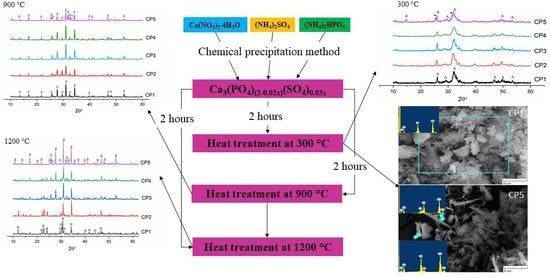
2.1. Powder Synthesis
2.2. Powder Characterisation
2.3. In Vitro Assays of Cytotoxicity and Cytocompatibility
2.3.1. The Indirect Contact Method
2.3.2. The Direct Contact Method
2.3.3. Cell Viability Assay
3. Results
3.1. Powder Chemical Composition
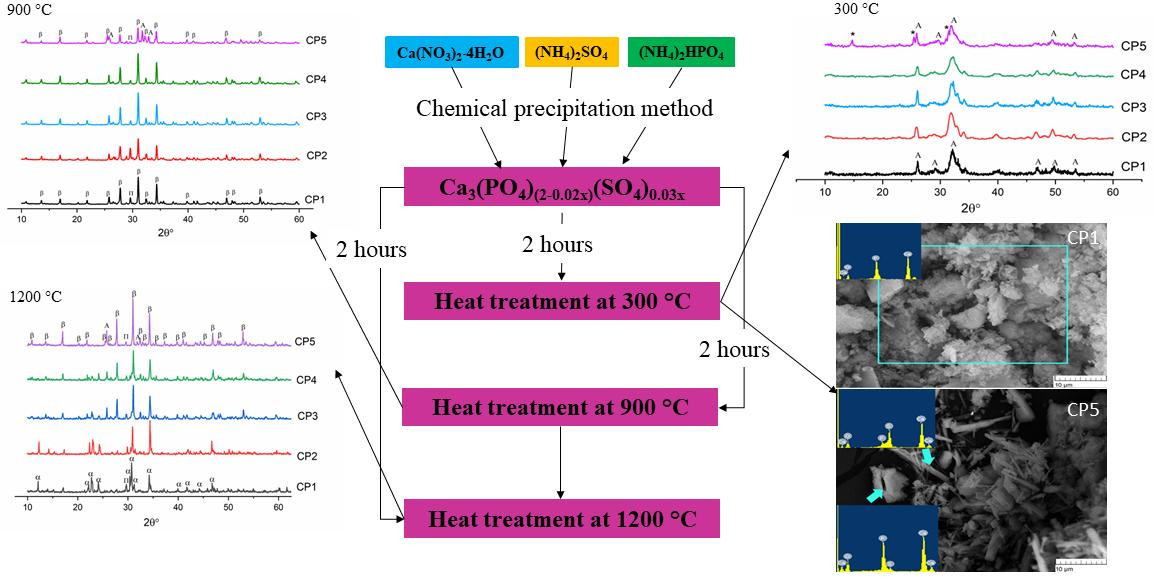
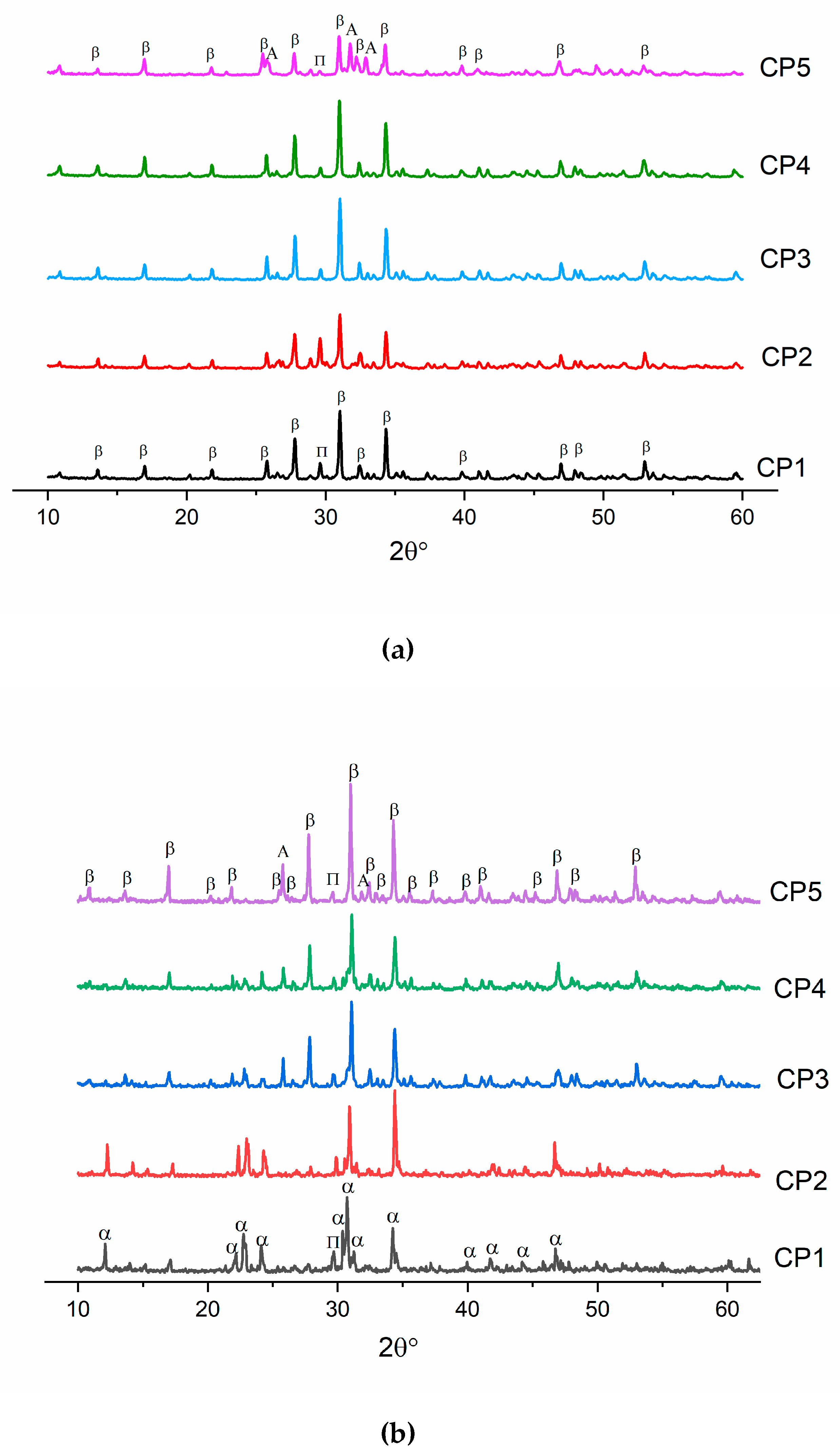
3.2. XRD Analysis
3.3. FTIR Analysis
3.4. Synchronous Thermal Analysis and Mass Spectra
3.5. Morphological Analyses by SEM
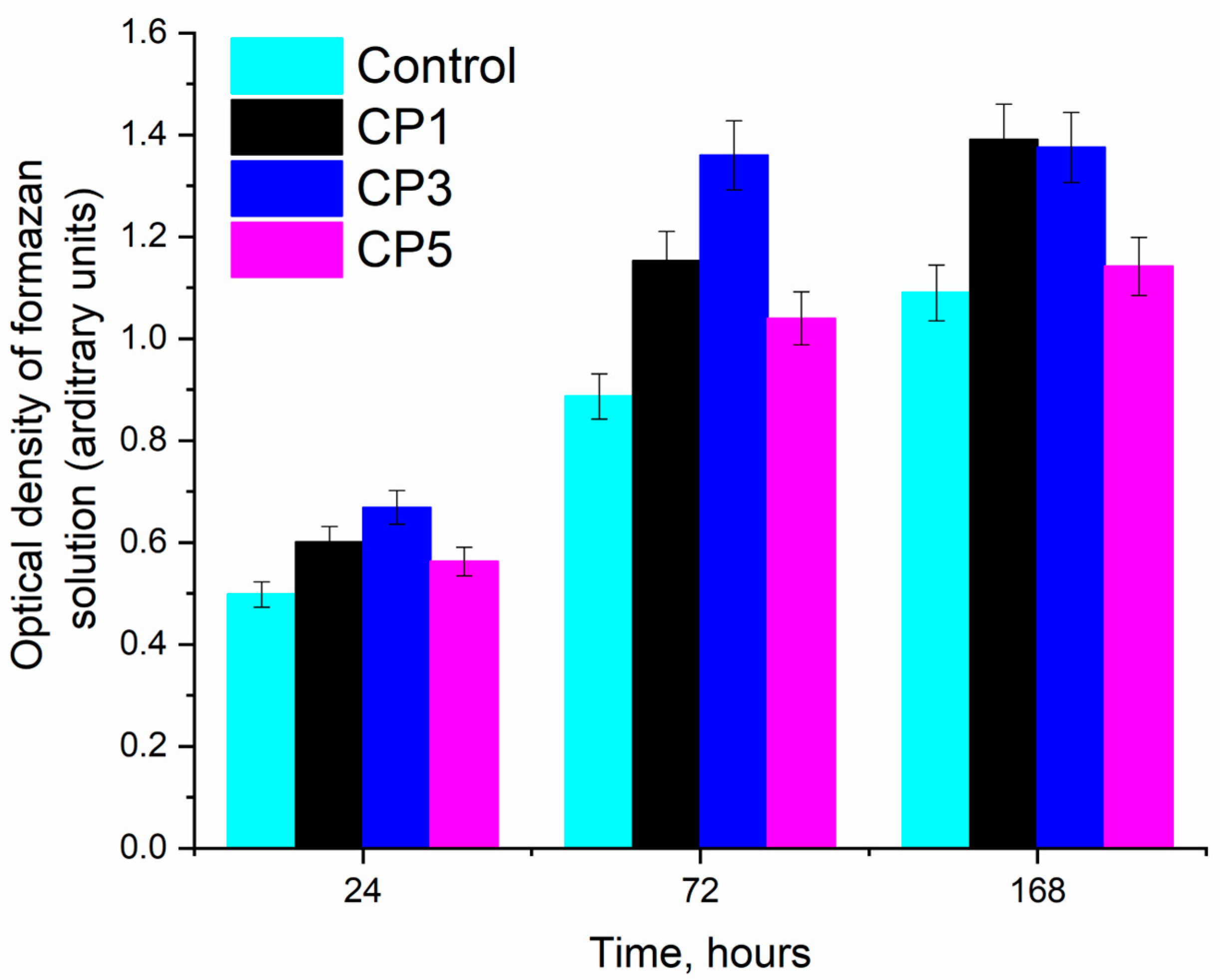
3.6. In Vitro Assays
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ruhé, P.; Wolke, J.; Spauwen, P.; Jansen, J. Calcium phosphate ceramics for bone tissue engineering. Eng. Bone 2005, 7–22. Available online: https://repository.ubn.ru.nl/bitstream/handle/2066/26979/26979_engibo.pdf?sequence=1#page=8 (accessed on 23 September 2005).
- Bohner, M.; Santoni, B.L.G.; Döbelin, N. β-tricalcium phosphate for bone substitution: Synthesis and properties. Acta Biomater. 2020, 113, 23–41. [Google Scholar] [CrossRef] [PubMed]
- Luna, M.C.; Jimenéz, E.B.M.; Horynová, M.; Gejdoš, P.; Klakurková, L.; de la Torre, S.D.; Kaiser, J.; Čelko, L. Tricalcium phosphate-magnesium interface: Microstructure and properties. Solid State Phenom. 2017, 258, 412–415. [Google Scholar] [CrossRef]
- Schmidleithner, C.; Malferrari, S.; Palgrave, R.; Bomze, D.; Schwentenwein, M.; Kalaskar, D.M. Application of high resolution DLP stereolithography for fabrication of tricalcium phosphate scaffolds for bone regeneration. Biomed. Mater. 2019, 14, 045018. [Google Scholar] [CrossRef]
- Fadeeva, I.V.; Goldberg, M.A.; Preobrazhensky, I.I.; Mamin, G.V.; Davidova, G.A.; Agafonova, N.V.; Fosca, M.; Russo, F.; Barinov, S.M.; Cavalu, S.; et al. Improved cytocompatibility and antibacterial properties of zinc-substituted brushite bone cement based on β-tricalcium phosphate. J. Mater. Sci. Mater. Med. 2021, 32, 99. [Google Scholar] [CrossRef]
- Antoniac, I.V.; Filipescu, M.; Barbaro, K.; Bonciu, A.; Birjega, R.; Cotrut, C.M.; Galvano, E.; Fosca, M.; Fadeeva, I.V.; Vadalà, G.; et al. Iron Ion-Doped Tricalcium Phosphate Coatings Improve the Properties of Biodegradable Magnesium Alloys for Biomedical Implant Application. Adv. Mater. Interfaces 2020, 7, 2000531. [Google Scholar] [CrossRef]
- Fadeeva, I.; Kalita, V.; Komlev, D.; Radiuk, A.; Fomin, A.; Davidova, G.; Fursova, N.; Murzakhanov, F.; Gafurov, M.; Fosca, M.; et al. In Vitro Properties of Manganese-Substituted Tricalcium Phosphate Coatings for Titanium Biomedical Implants Deposited by Arc Plasma. Materials 2020, 13, 4411. [Google Scholar] [CrossRef]
- Rahmanian, M.; Seyfoori, A.; Dehghan, M.M.; Eini, L.; Naghib, S.M.; Gholami, H.; Mohajeri, S.F.; Mamaghani, K.R.; Majidzadeh-A, K. Multifunctional gelatin–tricalcium phosphate porous nanocomposite scaffolds for tissue engineering and local drug delivery: In vitro and in vivo studies. J. Taiwan Inst. Chem. Eng. 2019, 101, 214–220. [Google Scholar] [CrossRef]
- Funayama, T.; Noguchi, H.; Kumagai, H.; Sato, K.; Yoshioka, T.; Yamazaki, M. Unidirectional porous beta-tricalcium phosphate and hydroxyapatite artificial bone: A review of experimental evaluations and clinical applications. J. Artif. Organs 2021, 24, 103–110. [Google Scholar] [CrossRef]
- Grigg, A.T.; Mee, M.; Mallinson, P.M.; Fong, S.K.; Gan, Z.; Dupree, R.; Holland, D. Cation substitution in β -tricalcium phosphate investigated using. J. Solid State Chem. 2014, 212, 227–236. [Google Scholar] [CrossRef]
- Vorndran, E.; Ewald, A.; Müller, F.A.; Zorn, K.; Kufner, A.; Gbureck, U. Formation and properties of magnesium–ammonium–phosphate hexahydrate biocements in the Ca–Mg–PO4 system. J. Mater. Sci. Mater. Med. 2011, 22, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Uskoković, V.; Graziani, V.; Wu, V.M.; Fadeeva, I.V.; Fomin, A.S.; Presniakov, I.A.; Fosca, M.; Ortenzi, M.; Caminiti, R.; Rau, J.V. Gold is for the mistress, silver for the maid: Enhanced mechanical properties, osteoinduction and antibacterial activity due to iron doping of tricalcium phosphate bone cements. Mater. Sci. Eng. C 2018, 94, 798–810. [Google Scholar] [CrossRef] [PubMed]
- Fadeeva, I.V.; Lazoryak, B.I.; Davidova, G.A.; Murzakhanov, F.F.; Gabbasov, B.F.; Petrakova, N.V.; Fosca, M.; Barinov, S.M.; Vadalà, G.; Uskoković, V.; et al. Antibacterial and cell-friendly copper-substituted tricalcium phosphate ceramics for biomedical implant applications. Mater. Sci. Eng. C 2021, 129, 112410. [Google Scholar] [CrossRef]
- Gokcekaya, O.; Ueda, K.; Ogasawara, K.; Kanetaka, H.; Narushima, T. In vitro evaluation of Ag-containing calcium phosphates: Effectiveness of Ag-incorporated β-tricalcium phosphate. Mater. Sci. Eng. C 2017, 75, 926–933. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, N.; Sato, K.; Yoshida, K.; Hashimoto, K.; Toda, Y. Preparation and characterization of β-tricalcium phosphate co-doped with monovalent and divalent antibacterial metal ions. Acta Biomater. 2009, 5, 3157–3164. [Google Scholar] [CrossRef]
- Kannan, S.; Goetz-Neunhoeffer, F.; Neubauer, J.; Ferreira, J.M.F. Ionic Substitutions in Biphasic Hydroxyapatite and β-Tricalcium Phosphate Mixtures: Structural Analysis by Rietveld Refinement. J. Am. Ceram. Soc. 2007, 91, 1–12. [Google Scholar] [CrossRef]
- Piccirillo, C.; Silva, M.; Pullar, R.; da Cruz, I.B.; Jorge, R.; Pintado, M.; Castro, P. Extraction and characterisation of apatite-and tricalcium phosphate-based materials from cod fish bones. Mater. Sci. Eng. C 2012, 33, 103–110. [Google Scholar] [CrossRef]
- Yin, X.; Stott, M.J. Theoretical insights into bone grafting silicon-stabilized α-tricalcium phosphate. J. Chem. Phys. 2005, 122, 024709. [Google Scholar] [CrossRef]
- Guo, H.; Wei, J.; Liu, C.S. Development of a degradable cement of calcium phosphate and calcium sulfate composite for bone reconstruction. Biomed. Mater. 2006, 1, 193–197. [Google Scholar] [CrossRef]
- Ene, R.; Nica, M.; Ene, D.; Cursaru, A.; Cirstoiu, C. Review of calcium-sulphate-based ceramics and synthetic bone substitutes used for antibiotic delivery in PJI and osteomyelitis treatment. EFORT Open Rev. 2021, 6, 297–304. [Google Scholar] [CrossRef]
- Walsh, W.R.; Morberg, P.; Yu, Y.; Yang, J.L.; Haggard, W.; Sheath, P.C.; Bruce, W.J.M. Response of a calcium sulfate bone graft substitute in a confined cancellous defect. Clin. Orthop. Relat. Res. 2003, 406, 228–236. [Google Scholar] [CrossRef]
- Yahav, A.; Kurtzman, G.M.; Katzap, M.; Dudek, D.; Baranes, D. Bone Regeneration Properties and Clinical Applications of Biphasic Calcium Sulfate. Dent. Clin. North Am. 2020, 64, 453–472. [Google Scholar] [CrossRef] [PubMed]
- Smirnov, V.V.; Khayrutdinova, D.R.; Antonova, O.S.; Goldberg, M.A.; Barinov, S.M. Bone Cements Based on Magnesium-Substituted Calcium Sulfates. Dokl. Chem. 2019, 485, 100–103. [Google Scholar] [CrossRef]
- Zima, A.; Paszkiewicz, Z.; Siek, D.; Czechowska, J.; Ślósarczyk, A. Study on the new bone cement based on calcium sulfate and Mg, CO3 doped hydroxyapatite. Ceram. Int. 2012, 38, 4935–4942. [Google Scholar] [CrossRef]
- Siek, D.; Czechowska, J.; Zima, A.; Ślósarczyk, A. Biodegradable cement type bone implant materials based on calcium phosphates and calcium sulphate. Eng. Biomater. 2015, 18, 2–6. [Google Scholar]
- Cai, Z.; Wu, Z.; Wan, Y.; Yu, T.; Zhou, C. Manipulation of the degradation behavior of calcium phosphate and calcium sulfate bone cement system by the addition of micro-nano calcium phosphate. Ceram. Int. 2021, 47, 29213–29224. [Google Scholar] [CrossRef]
- Baek, J.; Lee, H.; Jang, T.S.; Song, J.; Kim, H.E.; Jung, H. Incorporation of Calcium Sulfate Dihydrate into Hydroxyapatite Microspheres to Improve the Release of Bone Morphogenetic Protein-2 and Accelerate Bone Regeneration. ACS Biomater. Sci. Eng. 2018, 4, 846–856. [Google Scholar] [CrossRef]
- Jiang, N.; Dusane, D.H.; Brooks, J.R.; Delury, C.P.; Aiken, S.S.; Laycock, P.A.; Stoodley, P. Antibiotic loaded β-tricalcium phosphate/calcium sulfate for antimicrobial potency, prevention and killing efficacy of Pseudomonas aeruginosa and Staphylococcus aureus biofilms. Sci. Rep. 2021, 11, 1446. [Google Scholar] [CrossRef]
- Deyneko, D.V.; Titkov, V.V.; Fedyunin, F.D.; Spassky, D.A.; Volkov, S.N.; Borovikova, E.Y.; Lazoryak, B.I.; Aksenov, S.M. «Ellestadite»-type anionic [PO4]3– → [SO4]2– substitutions in β-Ca3(PO4)2 type compounds: A new route to design the inorganic phosphors. Ceram. Int. 2022, 48, 24012–24020. [Google Scholar] [CrossRef]
- Massit, A.; El Yacoubi, A.; Hmamouchi, S.; Rkhaila, A.; Boulouiz, A.; Ounine, K.; El Idrissi, B.C. “Sulphate-substituted tricalcium phosphate β-TCP: Effect of SO42− insertion and microwave conditions. Mater. Today Proc. 2022, 72, 3544–3549. [Google Scholar] [CrossRef]
- Sobczak-Kupiec, A.; Wzorek, Z. The influence of calcination parameters on free calcium oxide content in natural hydroxyapatite. Ceram. Int. 2012, 38, 641–647. [Google Scholar] [CrossRef]
- Liu, H.; Yazici, H.; Ergun, C.; Webster, T.J.; Bermek, H. An in vitro evaluation of the Ca/P ratio for the cytocompatibility of nano-to-micron particulate calcium phosphates for bone regeneration. Acta Biomater. 2008, 4, 1472–1479. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, N.; Yoshida, K.; Hashimoto, K.; Toda, Y. Thermal stability of β-tricalcium phosphate doped with monovalent metal ions. Mater. Res. Bull. 2009, 44, 1889–1894. [Google Scholar] [CrossRef]
- Somers, N.; Jean, F.; Lasgorceix, M.; Curto, H.; Urruth, G.; Thuault, A.; Petit, F.; Leriche, A. Influence of dopants on thermal stability and densification of β-tricalcium phosphate powders. Open Ceram. 2021, 7, 100168. [Google Scholar] [CrossRef]
- Abdel-Fattah, W.I.; Reicha, F.M.; Elkhooly, T.A. Nano-beta-tricalcium phosphates synthesis and biodegradation: 1. Effect of microwave and SO42- ions on β-TCP synthesis and its characterization. Biomed. Mater. 2008, 3, 034121. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Berry, E.E. Thermal Reactions between MI1 Sulphates and Acid Orthophosphates in the Solid State. J. Appl. Chem. Biotechnol. 1972, 22, 673–678. [Google Scholar] [CrossRef]
- Massit, A.; Fathi, M.; El Yacoubi, A.; Kholtei, A.; EL Idrissi, B.C. Effect of physical and chemical parameters on the β-Tricalcium phosphate synthesized by the wet chemical method. Mediterr. J. Chem. 2018, 7, 234–242. [Google Scholar] [CrossRef]
- Monma, H.; Moriyoshi, Y.; Ogata, H.; Okura, T. Characterization and hydration reactivity of α-tricaicium phosphate prepared by heat-ing with gypsum. Phosphorus Res. Bull. 2012, 27, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Choi, D.; Kumta, P.N. Mechano-chemical synthesis and characterization of nanostructured β- TCP powder. Mater. Sci. Eng. C 2007, 27, 377–381. [Google Scholar] [CrossRef]
- Sroka-Bartnicka, A.; Borkowski, L.; Ginalska, G.; Ślósarczyk, A.; Kazarian, S.G. Structural transformation of synthetic hydroxyapatite under simulated in vivo conditions studied with ATR-FTIR spectroscopic imaging. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2017, 171, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Ślósarczyk, A.; Paszkiewicz, Z.; Paluszkiewicz, C. FTIR and XRD evaluation of carbonated hydroxyapatite powders synthesized by wet methods. J. Mol. Struct. 2005, 744–747, 657–661. [Google Scholar] [CrossRef]
- Goldberg, M.; Gafurov, M.; Murzakhanov, F.; Fomin, A.; Antonova, O.; Khairutdinova, D.; Pyataev, A.; Makshakova, O.; Konovalov, A.; Leonov, A.; et al. Mesoporous iron(III)-doped hydroxyapatite nanopowders obtained via iron oxalate. Nanomaterials 2021, 11, 811. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Huang, R.; Qi, W.; Tong, L.; Su, R.; He, Z. Hydrolysis of cellulose by sulfonated magnetic reduced graphene oxide. Chem. Eng. J. 2015, 280, 90–98. [Google Scholar] [CrossRef]
- Ramesh, S.; Yuen, T.F.; Shen, C.J. Conductivity and FTIR studies on PEO–LiX [X: CF3SO3−, SO42−] polymer electrolytes. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2008, 69, 670–675. [Google Scholar] [CrossRef]
- Alshemary, Z.; Goh, Y.F.; Akram, M.; Razali, I.R.; Kadir, M.R.A.; Hussain, R. Microwave assisted synthesis of nano sized sulphate doped hydroxyapatite. Mater. Res. Bull. 2013, 48, 2106–2110. [Google Scholar] [CrossRef]
- Liang, C.; Li, Z.; Yang, D.; Li, Y.; Yang, Z.; Lu, W.W. Synthesis of calcium phosphate/calcium sulphate powder. Mater. Chem. Phys. 2004, 88, 285–289. [Google Scholar] [CrossRef]
- Tacker, R.C. Hydroxyl ordering in igneous apatite. Am. Miner. 2004, 89, 1411–1421. [Google Scholar] [CrossRef]
- Sinusaite, L.; Grigoraviciute-Puroniene, I.; Popov, A.; Ishikawa, K.; Kareiva, A.; Zarkov, A. Controllable synthesis of tricalcium phosphate (TCP) polymorphs by wet precipitation: Effect of washing procedure. Ceram. Int. 2019, 45, 12423–12428. [Google Scholar] [CrossRef]
- Safronova, T.V.; Putlyaev, V.I.; Kurbatova, S.A.; Shatalova, T.; Larionov, D.S.; Kozlov, D.A.; Evdokimov, P.V. Properties of amorphous calcium pyrophosphate powder synthesized via ion exchange for the preparation of bioceramics. Inorg. Mater. 2015, 51, 1177–1184. [Google Scholar] [CrossRef]
- Ryu, H.-S.; Youn, H.-J.; Hong, K.S.; Chang, B.-S.; Lee, C.-K.; Chung, S.-S. An improvement in sintering property of b-tricalcium phosphate by addition of calcium pyrophosphate. Biomaterials 2002, 23, 909–914. [Google Scholar] [CrossRef] [PubMed]
- Nazir, R.; Khan, A.S.; Ahmed, A.; Ur-Rehman, A.; Chaudhry, A.A.; Rehman, I.U.; Wong, F.S. Synthesis and in-vitro cytotoxicity analysis of microwave irradiated nano-apatites. Ceram. Int. 2012, 39, 4339–4347. [Google Scholar] [CrossRef]
- Urban, R.M.; Turner, T.M.; Hall, D.J.; Inoue, N.; Gitelis, S. Increased bone formation using calcium sulfate-calcium phosphate composite graft. Clin. Orthop. Relat. Res. 2007, 459, 110–117. [Google Scholar] [CrossRef]
- Dong, Z.; Saikumar, P.; Weinberg, J.M.; Venkatachalam, M.A. Calcium in cell injury and death. Annu. Rev. Pathol. Mech. Dis. 2006, 1, 405–434. [Google Scholar] [CrossRef] [PubMed]
- Fadeeva, I.; Gafurov, M.; Kiiaeva, I.; Orlinskii, S.; Kuznetsova, L.; Filippov, Y.; Fomin, A.; Davydova, G.; Selezneva, I.; Barinov, S. Tricalcium Phosphate Ceramics Doped with Silver, Copper, Zinc, and Iron (III) Ions in Concentrations of Less Than 0.5 wt.% for Bone Tissue Regeneration. Bionanoscience 2016, 7, 434–438. [Google Scholar] [CrossRef]
- Matsunaga, K.; Kubota, T.; Toyoura, K.; Nakamura, A. First-principles calculations of divalent substitution of Ca2+ in tricalcium phosphates. Acta Biomaterialia 2015, 23, 329–337. [Google Scholar] [CrossRef]
- Zhang, Z.; Niu, J.; Zhou, W.; Xu, D.; Du, H. Samarium doped apatite-type orange-red emitting phosphor Ca5(PO4)2SiO4 with satisfactory thermal properties for n-UV w-LEDs. J. Rare Earths 2019, 37, 949–954. [Google Scholar] [CrossRef]
- Šupová, M. Substituted hydroxyapatites for biomedical applications: A review. Ceram. Int. 2015, 41, 9203–9231. [Google Scholar] [CrossRef]
- Smirnov, S.; Khayrutdinova, D.R.; Antonova, O.S.; Golgberg, M.A.; Barinov, S.M. The effect of phosphate group replacement by sulfate groups on the phase formation in the synthesis of hydroxyapatite. Dokl. Chem. 2017, 476, 223–225. [Google Scholar] [CrossRef]
- Marraha, M.; Heughebaert, J.C. Preparation et etude physico-chimique d’une serie de phosphosulfates de calcium Ca21-x(PO4)14-2x(SO4)2x (formula omitted) presentant la structure de l’orthophosphate tricalcique anhydre β-Ca3(PO4)2. Phosphorus Sulfur Silicon Relat. Elements 1989, 44, 177–192. [Google Scholar] [CrossRef]
- Marraha, M.; Heughebaert, J.-C.; Bonel, G. Formation of a new solid β tricalcium orthophosphate structure, from reaction between hydroxyapatite and ammonium sulfate. Ceram. Int. 1983, 9, 93–96. [Google Scholar] [CrossRef]
- Sabouri, Z.; Mostafapour, A.; Darroudi, M. Biopolymer-template synthesized CaSO4 nanoparticles and evaluation of their photocatalytic activity and cytotoxicity effects ‘Green synthesis of medicinal nanoparticles and investigation of their cytotoxicity effects’ View project Fe3O4 Nanoparticles, Magnetic nanocarriers, drug delivery, colon cancer, oxaliplatin View project. Ceram. Int. 2022, 48, 16306–16311. [Google Scholar] [CrossRef]
- Radha, G.; Venkatesan, B.; Jaisankar, S.N.; Rajashree, P.; Balakumar, S. Interplay between surface chemistry and osteogenic behaviour of sulphate substituted nano-hydroxyapatite. Mater. Sci. Eng. C 2020, 120, 111617. [Google Scholar] [CrossRef]
- Hall, D.J.; Turner, T.M.; Urban, R.M. Healing bone lesion defects using injectable CaSO4/CaPO4-TCP bone graft substitute compared to cancellous allograft bone chips in a canine model. J. Biomed. Mater. Res. Part B Appl. Biomater. 2018, 107, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Urban, R.M.; Turner, T.M.; Hall, D.J.; Inoue, N.; Gitelis, S. Advanced Bone Regeneration Using an Injectable CaSO4/CaPO4-TCP Composite Compared to Cancellous Bone Autograft in a Canine Model. In Proceedings of the 54th Annual Meeting of the Orthopaedic Research Society, San Francisco, CA, USA, 2–5 March 2008. No. 67. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Content of Sulphate Groups, mol.% (x) | Calculated Formula | mCa(NO3)2·4H2O, g | m(NH4)2SO4, g | m(NH4)2HPO4, g |
|---|---|---|---|---|---|
| CP1 | 0 | Ca3(PO4)2 | 114.00 | - | 43.00 |
| CP2 | 1.5 | Ca3(PO4)1.97(SO4)0.045 | 113.70 | 0.95 | 41.70 |
| CP3 | 3.5 | Ca3(PO4)1.93(SO4)0.105 | 112.90 | 2.20 | 40.60 |
| CP4 | 7.0 | Ca3(PO4)1.86(SO4)0.21 | 111.70 | 4.40 | 38.70 |
| CP5 | 12.0 | Ca3(PO4)1.76(SO4)0.36 | 110.00 | 7.40 | 36.01 |
| Sample ID | Content of Sulphate Groups, mol.% | Amount of Sulphur (300 °C), wt.% | SSA, m2/g | |
|---|---|---|---|---|
| Theoretical | Experimental * | |||
| CP1 | 0.0 | 0.00 | 0.00 | 95.1 ± 0.1 |
| CP2 | 1.5 | 0.49 | 0.42 | 81.7 ± 0.1 |
| CP3 | 3.5 | 1.10 | 1.00 | 92.8 ± 0.1 |
| CP4 | 7.0 | 2.10 | 2.00 | 96.5 ± 0.1 |
| CP5 | 12.0 | 3.58 | 3.48 | 87.6 ± 0.1 |
| Sample ID | 900 °C | 1200 °C | ||
|---|---|---|---|---|
| Measured Amount of Sulphur *, wt.% | Calculated Content of Sulphate Groups, mol.% | Measured Amount of Sulphur *, wt.% | Calculated Content of Sulphate Groups, mol.% | |
| CP1 | 0.000 | 0.000 0.057 0.080 3.600 11.010 | 0.000 | 0.000 |
| CP2 | 0.019 | 0.005 | 0.015 | |
| CP3 | 0.030 | 0.006 | 0.017 | |
| CP4 | 1.200 | 0.188 | 0.560 | |
| CP5 | 3.670 | 1.960 | 5.880 | |
| Sample ID | a, nm | c, nm | V, nm3 | D, nm |
|---|---|---|---|---|
| Theoretical * | 0.1043 | 0.3737 | 0.3527 | - |
| CP1 | 0.1043(3) | 0.3734(3) | 0.3521 | 43 |
| CP2 | 0.1043(1) | 0.3735(5) | 0.3519 | 43 |
| CP3 | 0.1042(3) | 0.3732(1) | 0.3511 | 33 |
| CP4 | 0.1043(3) | 0.3739(1) | 0.3524 | 38 |
| CP5 | 0.1045(1) | 0.3741(1) | 0.3538 | 44 |
| Sample ID | pH | OD of Formazan Solution (Conventional Arbitrary Units) | PVC (%) | Toxicity Index (%) |
|---|---|---|---|---|
| Positive control-DMSO-20% | 7.4 | 0.062 ± 0.02 | 10.7 | 89.3 |
| Negative control-CGM | 7.4 | 0.578 ± 0.01 | 100.0 | 0.0 |
| CP1 | 7.8 | 0.457 ± 0.01 | 79.1 | 20.9 |
| CP3 | 7.5 | 0.528 ± 0.02 | 91.3 | 8.7 |
| CP5 | 7.5 | 0.485 ± 0.03 | 83.9 | 16.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khayrutdinova, D.R.; Goldberg, M.A.; Antonova, O.S.; Krokhicheva, P.A.; Fomin, A.S.; Obolkina, T.O.; Konovalov, A.A.; Akhmedova, S.A.; Sviridova, I.K.; Kirsanova, V.A.; et al. Effects of Heat Treatment on Phase Formation in Cytocompatible Sulphate-Containing Tricalcium Phosphate Materials. Minerals 2023, 13, 147. https://doi.org/10.3390/min13020147
Khayrutdinova DR, Goldberg MA, Antonova OS, Krokhicheva PA, Fomin AS, Obolkina TO, Konovalov AA, Akhmedova SA, Sviridova IK, Kirsanova VA, et al. Effects of Heat Treatment on Phase Formation in Cytocompatible Sulphate-Containing Tricalcium Phosphate Materials. Minerals. 2023; 13(2):147. https://doi.org/10.3390/min13020147
Chicago/Turabian StyleKhayrutdinova, Dinara R., Margarita A. Goldberg, Olga S. Antonova, Polina A. Krokhicheva, Alexander S. Fomin, Tatiana O. Obolkina, Anatoliy A. Konovalov, Suraya A. Akhmedova, Irina K. Sviridova, Valentina A. Kirsanova, and et al. 2023. "Effects of Heat Treatment on Phase Formation in Cytocompatible Sulphate-Containing Tricalcium Phosphate Materials" Minerals 13, no. 2: 147. https://doi.org/10.3390/min13020147