In Situ Hyperspectral Raman Imaging of Ternesite Formation and Decomposition at High Temperatures


Abstract
:1. Introduction
2. Materials and Methods
2.1. Characterization of the Starting Materials and Sample Preparation
2.2. Experimental Details
2.3. Raman Spectroscopy
2.3.1. High-Temperature Raman Spectroscopy of Ternesite
2.3.2. Hyperspectral Raman Imaging
3. Results
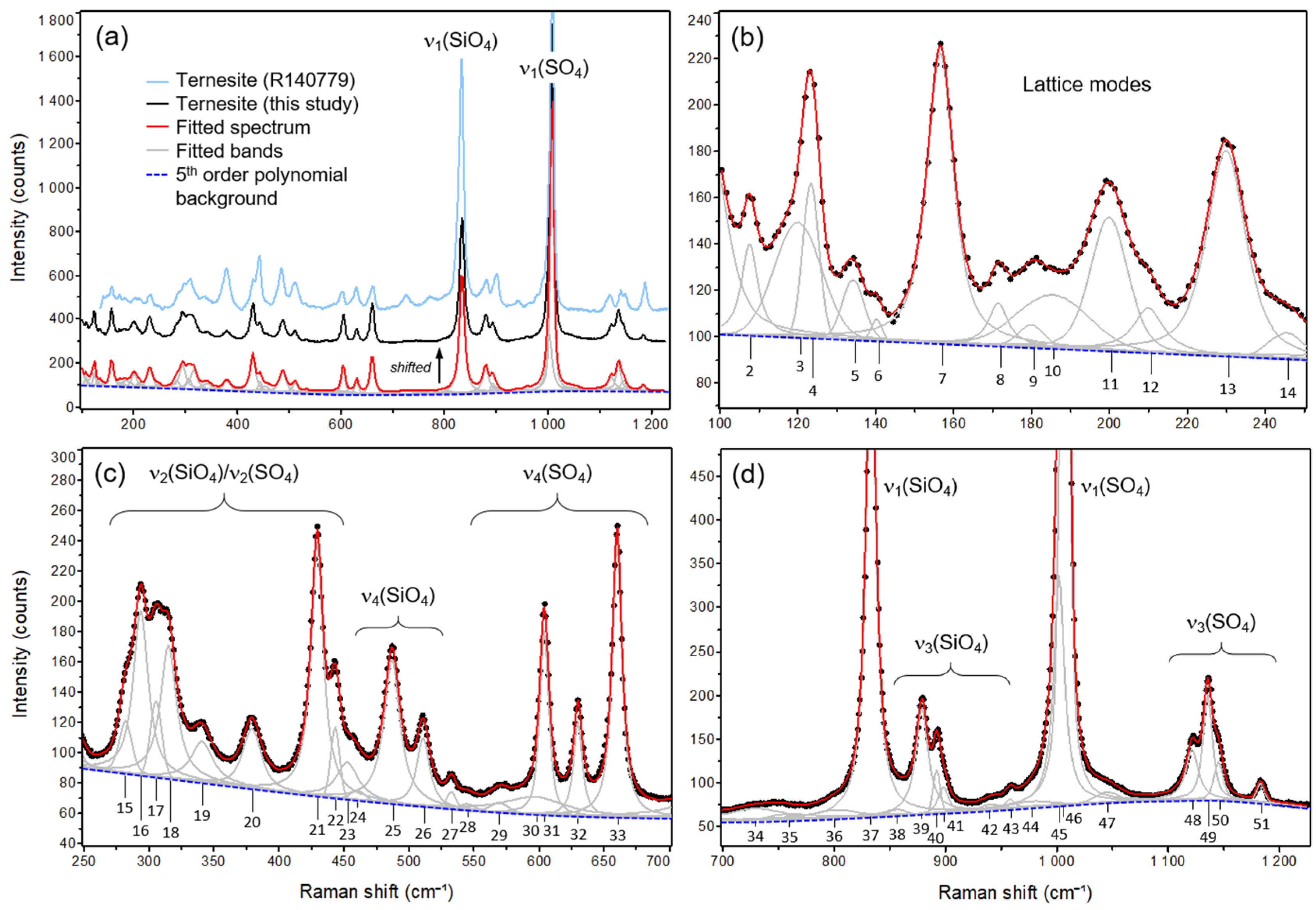
3.1. Room Temperature (RT) Raman Spectrum
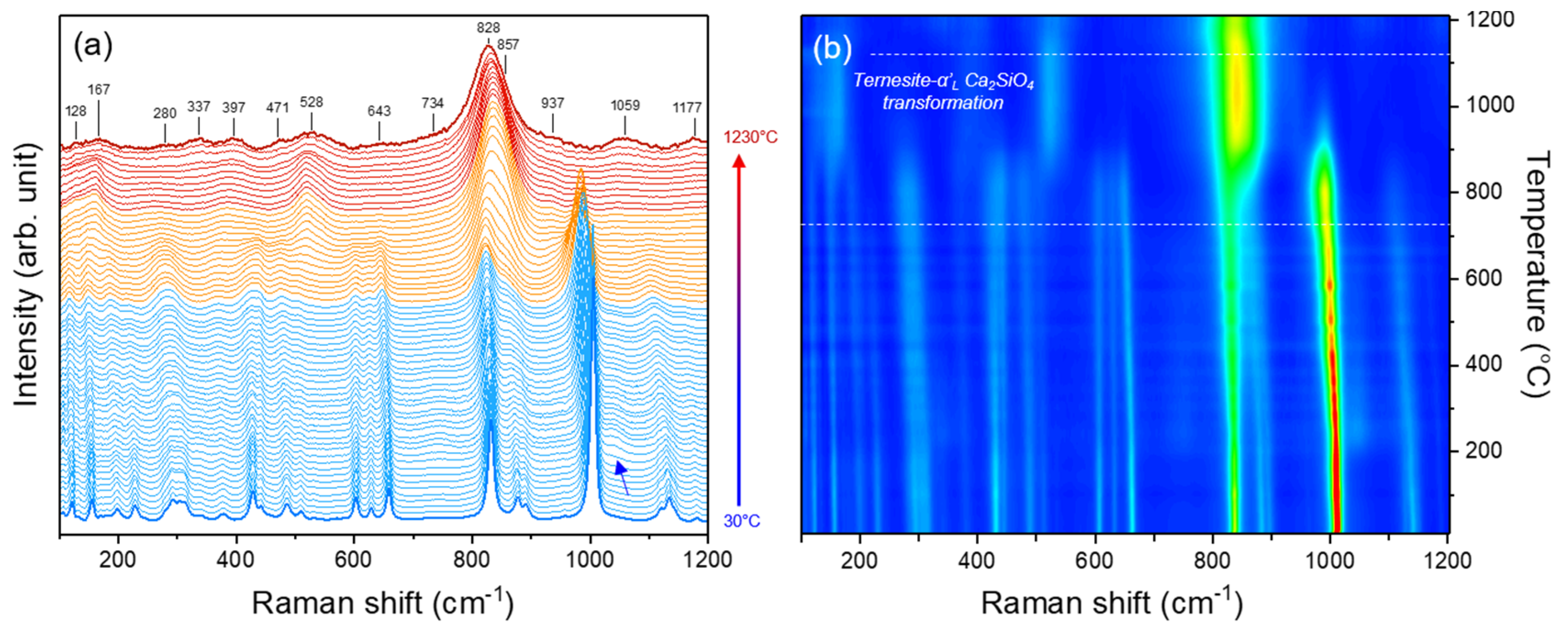
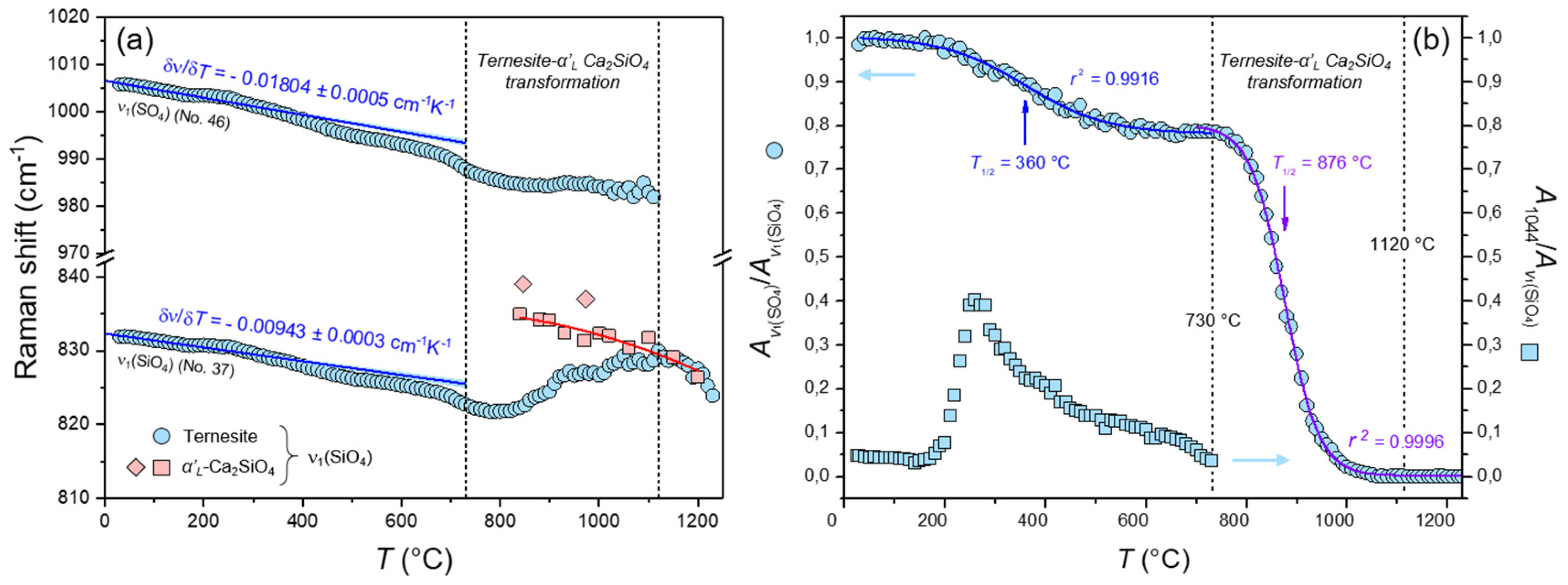
3.2. High-Temperature (HT) Raman Spectra of Ternesite
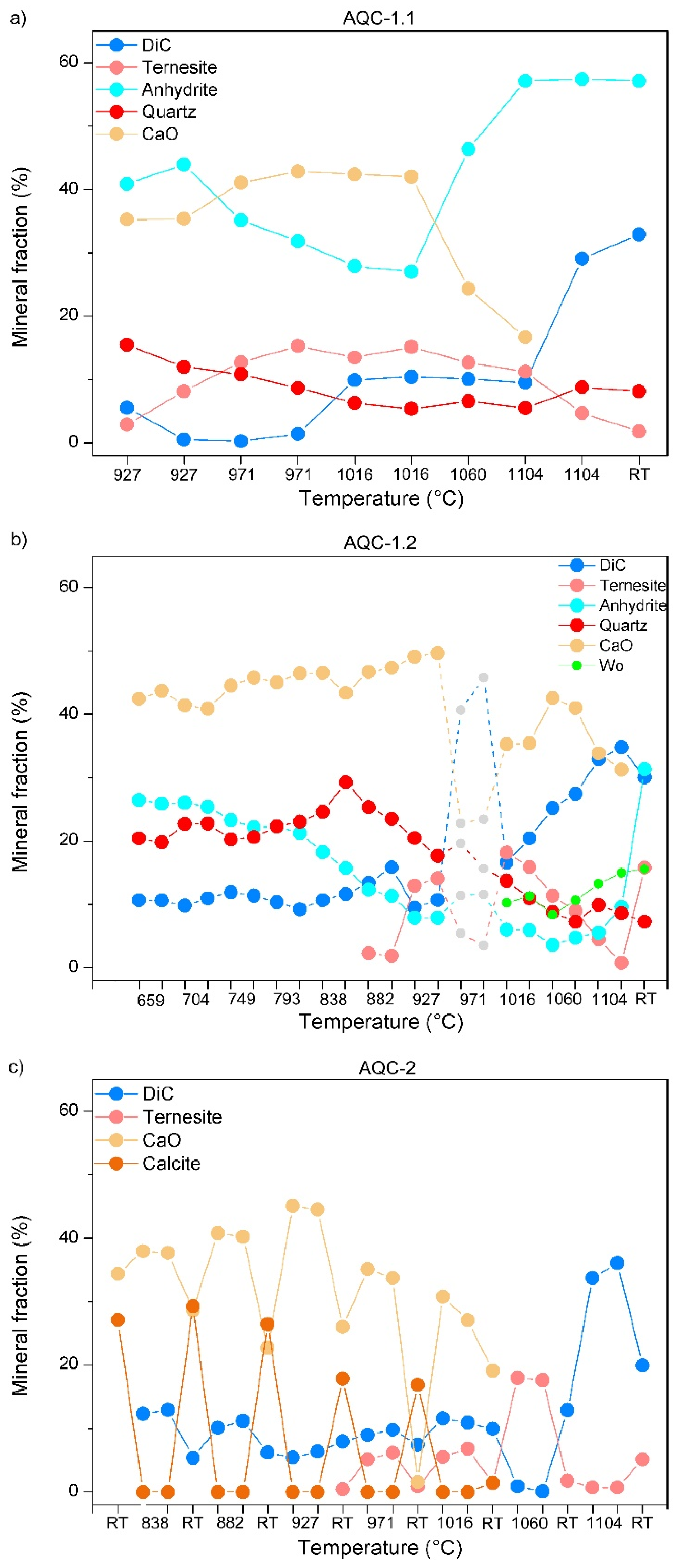
3.3. In Situ Experiments AQC-1.1 and AQC-1.2
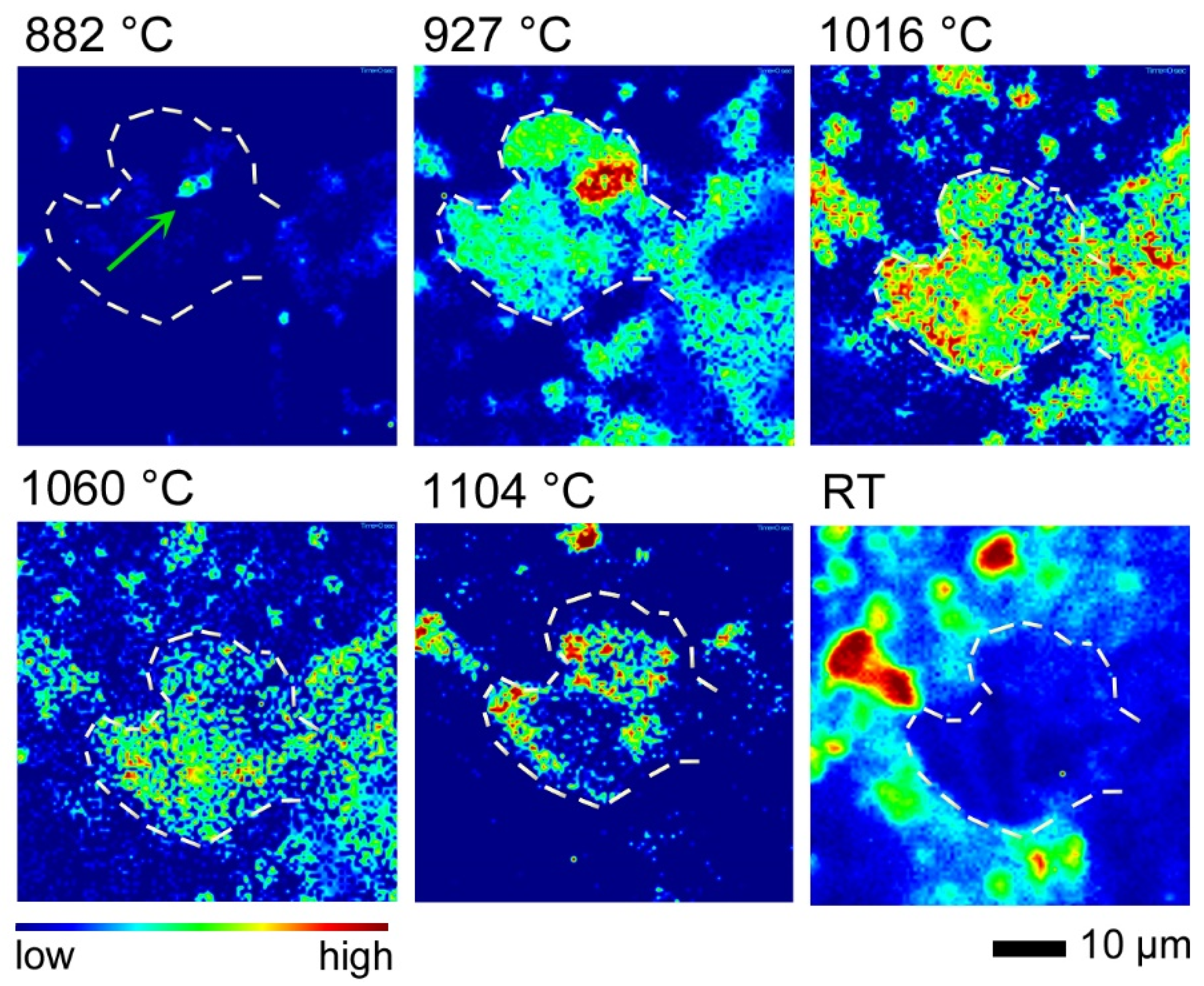
3.4. In Situ/Quench Experiment AQC-2
4. Discussion
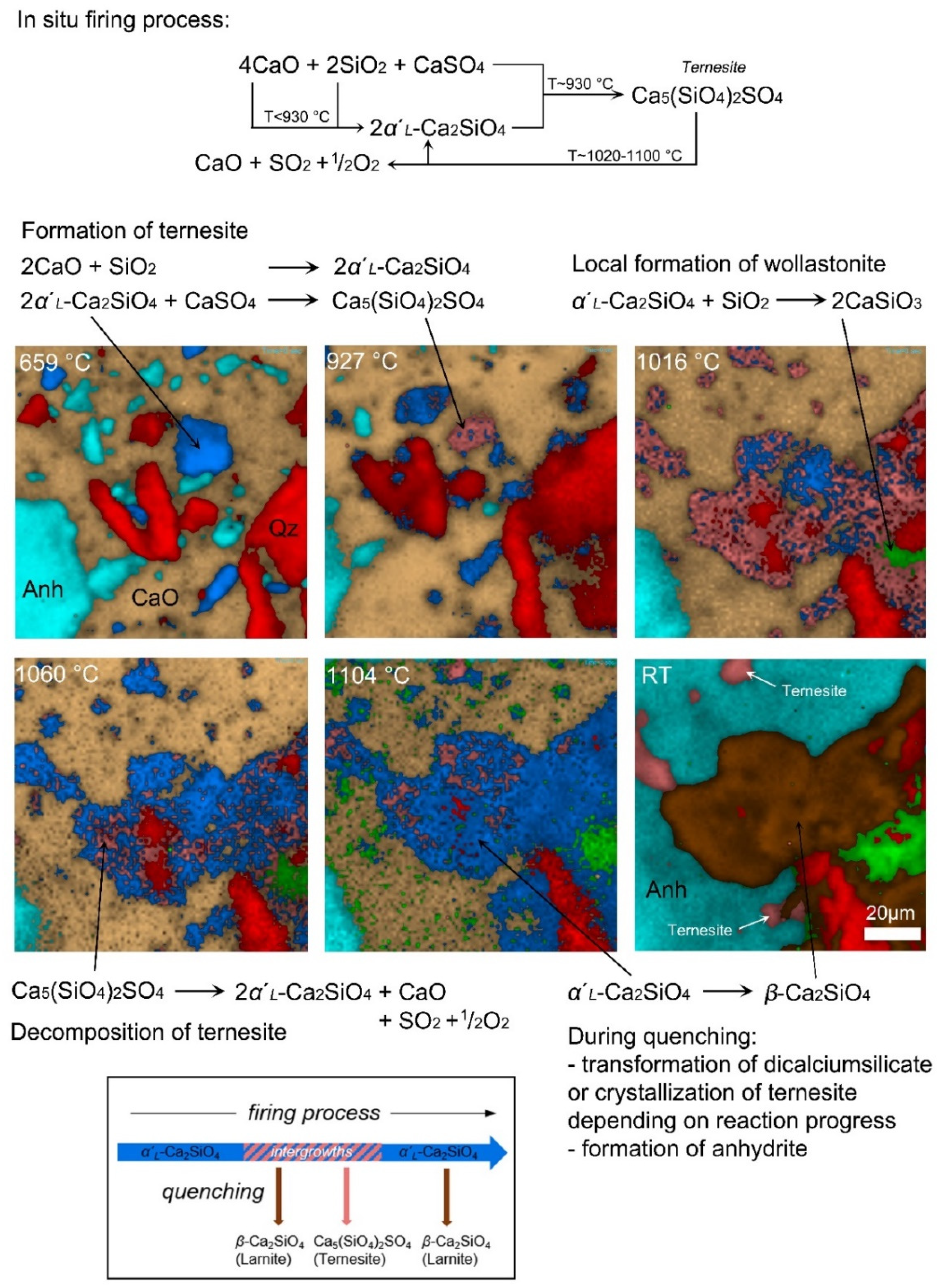
4.1. The Formation of Calcium Silicates
4.2. The Formation and Decomposition of Ternesite
4.3. The Ternesite–Dicalcium Silicate Co-Existence
5. Summary and Conclusion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Irran, E.; Tillmanns, E.; Hentschel, G. Ternesite, Ca5(SiO4)2SO4, a new mineral from the Ettringer Bellerberg/Eifel, Germany. Miner. Pet. 1997, 60, 121–132. [Google Scholar] [CrossRef]
- Galuskin, E.; Galuskina, I.; Gfeller, F.; Krüger, B.; Kusz, J.; Vapnik, Y.; Dulski, M.; Dzierżanowski, P. Silicocarnotite, Ca5[(SiO4)(PO4)](PO4), a new old mineral from the Negev Desert, Israel, and the ternesite–silicocarnotite solid solution: Indicators of high-temperature alteration of pyrometamorphic rocks of the Hatrurim Complex, Southern Levant. Eur. J. Miner. 2016, 28, 105–123. [Google Scholar] [CrossRef]
- Hanein, T.; Galan, I.; Glasser, F.P.; Skalamprinos, S.; Elhoweris, A.; Imbabi, M.S.; Bannerman, M.N. Stability of ternesite and the production at scale of ternesite-based clinkers. Cem. Concr. Res. 2017, 98, 91–100. [Google Scholar] [CrossRef]
- Brotherton, P.D.; Epstein, J.M.; Pryce, M.W.; White, A.H. Crystal Structure of “Calcium Sulphosilicate”, Ca5(SiO4)2SO4. Aust. J. Chem. 1974, 27, 657–660. [Google Scholar] [CrossRef]
- Pryce, M.W. Calcium sulphosilicate in lime-kiln wall coating. Miner. Mag. 1972, 38, 968–971. [Google Scholar] [CrossRef]
- Neuroth, M. Internal Report of RWE, KW Neurath, Unpublished work. 2017.
- Gutt, W.; Smith, M.A. A new calcium silicosulphate. Nature 1966, 210, 408–409. [Google Scholar] [CrossRef]
- Dienemann, W.; Schmitt, D.; Bullerjahn, F.; Haha, M. Ben Belite-Calciumsulfoaluminate-Ternesite (BCT)—A new low-carbon clinker technology. Cem. Int. 2013, 11, 100–109. [Google Scholar]
- Bullerjahn, F.; Schmitt, D.; Haha, M. Ben Cement and Concrete Research Effect of raw mix design and of clinkering process on the formation and mineralogical composition of (ternesite) belite calcium sulphoaluminate ferrite clinker. Cem. Concr. Res. 2014, 59, 87–95. [Google Scholar] [CrossRef]
- Shen, Y.; Qian, J.; Huang, Y.; Yang, D. Synthesis of belite sulfoaluminate-ternesite cements with phosphogypsum. Cem. Concr. Compos. 2015, 63, 67–75. [Google Scholar] [CrossRef]
- Galan, I.; Elhoweris, A.; Hanein, T.; Bannerman, M.N.; Glasser, F.P. Advances in clinkering technology of calcium sulfoaluminate cement. Adv. Cem. Res. 2017, 1–13. [Google Scholar] [CrossRef]
- Hanein, T.; Elhoweris, A.; Khare, S.; Skalamprinos, S.; Jen, G.; Whittaker, M.; Imbabi, M.S.; Glasser, F.P.; Galan, I.; Bannerman, M.N. Production of belite calcium sulfoaluminate cement using sulfur as a fuel and as a source of clinker sulfur trioxide: Pilot kiln trial. Adv. Cem. Res. 2016, 28, 643–653. [Google Scholar] [CrossRef] [Green Version]
- Hofman, H.; Mostowitsch, W. The behavior of calcium sulfate at elevated temperatures with some fluxes. Trans. Am. Inst. Min. Metall. Eng. 1909, 32, 628–653. [Google Scholar]
- Mihara, N.; Kuchar, D.; Kojima, Y.; Matsuda, H. Reductive decomposition of waste gypsum with SiO2, Al2O3, and Fe2O3 additives. J. Mater. Cycles Waste Manag. 2007, 9, 21–26. [Google Scholar] [CrossRef]
- Böhme, N.; Hauke, K.; Neuroth, M.; Geisler, T. In situ Raman imaging of high-temperature solid-state reactions in the CaSO4–SiO2 system. Int. J. Coal Sci. Technol. 2019, 6, 247–259. [Google Scholar] [CrossRef] [Green Version]
- Hauke, K.; Kehren, J.; Böhme, N.; Zimmer, S.; Geisler, T. In situ Hyperspectral Raman Imaging: A new Method to investigate Sintering Processes of Ceramic Material at High Temperature. Appl. Sci. 2019, 9, 1310. [Google Scholar] [CrossRef] [Green Version]
- Stange, K.; Lenting, C.; Geisler, T. Insights into the evolution of carbonate-bearing kaolin during sintering revealed by in situ hyperspectral Raman imaging. J. Am. Ceram. Soc. 2018, 101, 897–910. [Google Scholar] [CrossRef]
- Lafuente, B.; Downs, R.T.; Yang, H.; Stone, N. The power of databases: The RRUFF project. In Highlights in Mineralogical Crystallography; Armbruster, T., Danisi, R.M., Eds.; De Gruyter: Berlin, Germany, 2015; pp. 1–30. [Google Scholar]
- Yuan, X.; Mayanovic, R.A.; Zheng, H. Determination of pressure from measured Raman frequency shifts of anhydrite and its application in fluid inclusions and HDAC experiments. Geochim. Cosmochim. Acta 2016, 194, 253–265. [Google Scholar] [CrossRef]
- Remy, C.; Reynard, B.; Madon, M. Raman Spectroscopic Investigations of Dicalcium Silicate: Polymorphs and High-Temperature Phase Transformations. J. Am. Ceram. Soc. 1997, 80, 413–423. [Google Scholar] [CrossRef]
- Ben Mabrouk, K.; Kauffmann, T.H.; Aroui, H.; Raman, M.D.F. Raman study of cation effect on sulfate vibration modes in solid state and in aqueous solutions. J. Raman Spectrosc. 2018, 44, 1603–1608. [Google Scholar] [CrossRef]
- Reynard, B.; Remy, C.; Takir, F. High-pressure Raman spectroscopic study of Mn2GeO4, Ca2GeO4, Ca2SiO4, and CaMgGeO4 olivines. Phys. Chem. Miner. 1997, 24, 77–84. [Google Scholar] [CrossRef]
- Mitsuda, T.; Asami, J.; Matsubara, Y.; Toraya, H. Hydrothermal formation of γ-dicalcium silicate from lime-silica mixt.pdf. Cem. Concr. Res. 1985, 15, 613–621. [Google Scholar] [CrossRef]
- Singh, N.B. Hydrothermal synthesis of β-dicalcium silicate (β-Ca2SiO4). Prog. Cryst. Growth Charact. Mater. 2006, 52, 77–83. [Google Scholar] [CrossRef]
- Fierens, P.; Picquet, P. Kinetic Studies of the Thermal Synthesis of Calcium Silicates above 1400 °C: I, Dynamic Thermal Synthesis of Ca2SiO4. J. Am. Ceram. Soc. 1975, 58, 50–51. [Google Scholar] [CrossRef]
- Kostakis, G. Mineralogical composition of boiler fouling and slagging deposits and their relation to fly ashes: The case of Kardia power plant. J. Hazard. Mater. 2011, 185, 1012–1018. [Google Scholar] [CrossRef]
- Li, J.; Zhu, M.; Zhang, Z.; Zhang, K.; Shen, G.; Zhang, D. The mineralogy, morphology and sintering characteristics of ash deposits on a probe at different temperatures during combustion of blends of Zhundong lignite and a bituminous coal in a drop tube furnace. Fuel Process. Technol. 2016, 149, 176–186. [Google Scholar] [CrossRef]
- Tschegg, C.; Ntaflos, T.; Hein, I. Thermally triggered two-stage reaction of carbonates and clay during ceramic firing-A case study on Bronze Age Cypriot ceramics. Appl. Clay Sci. 2009, 43, 69–78. [Google Scholar] [CrossRef]
- Ptáček, P.; Opravil, T.; Šoukal, F.; Havlica, J.; Holešinský, R. Kinetics and mechanism of formation of gehlenite, Al-Si spinel and anorthite from the mixture of kaolinite and calcite. Solid State Sci. 2013, 26, 53–58. [Google Scholar] [CrossRef]
- Rashid, A.R.; Shamsudin, R.; Abdul Hamid, M.A.; Jalar, A. Low temperature production of wollastonite from limestone and silica sand through solid-state reaction. J. Asian Ceram. Soc. 2014, 2, 77–81. [Google Scholar] [CrossRef] [Green Version]
- Choi, G.; Glasser, F.P.; Skalny, C.J. The sulphur cycle in cement kilns: Vapour pressures and solid-phase stability of the sulphate phases. Cem. Concr. Res. 1988, 18, 367–374. [Google Scholar] [CrossRef]
- Scholten, T. Reaktionskinetik von sulfatischen Klinkerphasen in Zementen mit verminderter CO2-Last. Ph.D. Dissertation, Technische Universität Clausthal, Clausthal-Zellerfeld, Germany, 2017. [Google Scholar]
- Rheingans, B.F. Analysis of the kinetics of phase transformations. J. Mater. Sci. 1992, 27, 3977–3987. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Oxides | Range [1] (wt.%) | Average [1] (wt.%) | Range [This Study] (wt.%) | Average [This Study] (wt.%) |
|---|---|---|---|---|
| CaO | 57.32–60.35 | 58.90 ± 0.83 | 57.69–59.21 | 58.50 ± 0.28 |
| SiO2 | 23.05–27.23 | 25.22 ± 1.28 | 24.57–25.79 | 25.13 ± 0.28 |
| SO3 | 13.80–18.04 | 16.34 ± 1.25 | 15.21–17.02 | 16.27 ± 0.34 |
| Exp. | Anh:Qtz:CaO (mol ratio) | Type of Experiment | Trange | Tsteps | Trate | Acquisition | Recording |
|---|---|---|---|---|---|---|---|
| (°C) | (°C) | (°C/min) | Time (s) | Time (h) | |||
| Ternesite | in situ | 30–1230 | 10 | 10 ↗↘ | 100 s | − | |
| AQC-1.1 | 1:2:4 | in situ | 927–1104 | ~50 | 10 ↗↘ | 0.5 | 2 × 2 (4) * |
| AQC-1.2 | 1:2:4 | in situ | 659–1104 | ~50 | 10 ↗↘ | 0.5 | 2 × 2 (4) * |
| AQC-2 | 1:2:4 | in situ/quench | 838–1104 | ~50 | 10 ↗↘ | 0.5 | 2 × 2 (4) * |
| No. | Raman Shift | FWHM | Relative Intensity * | Remarks | Herzberg Numbering | Approximate Type of Mode |
|---|---|---|---|---|---|---|
| (cm−1) | (cm−1) | |||||
| 1 | 95.4 | 11.1 | vw | outside measured frequency range, but used in the fitting process | Lattice vibrations | |
| 2 | 106.6 | 5.0 | vw | |||
| 3 | 119.0 | 14.3 | vw | |||
| 4 | 122.3 | 4.9 | w | |||
| 5 | 133.1 | 6.6 | vw | |||
| 6 | 139.2 | 3.2 | vw | |||
| 7 | 155.5 | 8.2 | s | |||
| 8 | 170.5 | 5.8 | vw | |||
| 9 | 179.0 | 7.7 | vw | |||
| 10 | 184.4 | 21.9 | vw | |||
| 11 | 198.9 | 11.4 | w | |||
| 12 | 209.0 | 9.2 | vw | |||
| 13 | 229.0 | 12.0 | s | |||
| 14 | 244.5 | 11.1 | vw | |||
| 15 | 280.9 | 11.6 | w | v2(SO4) or v2(SiO4) | O–S–O or O–Si–O symmetric bending | |
| 16 | 292.6 | 13.8 | s | |||
| 17 | 304.4 | 10.4 | w | |||
| 18 | 313.9 | 14.8 | s | |||
| 19 | 339.8 | 20.1 | vw | |||
| 20 | 378.1 | 17.1 | w | |||
| 21 | 428.2 | 11.4 | s | v2(SO4) | O–S–O symmetric bending | |
| 22 | 442.1 | 8.0 | w | |||
| 23 | 451.5 | 20.1 | w | v2(SO4) or v2(SiO4) | O–S–O or O–Si–O symmetric bending | |
| 24 | 459.6 | 13.6 | vw | |||
| 25 | 485.8 | 14.7 | s | |||
| 26 | 509.7 | 11.9 | w | v4(SiO4) | O–Si–O anti-symmetric bending | |
| 27 | 531.4 | 8.2 | w | |||
| 28 | 543.6 | 15.1 | vw | ? | ||
| 29 | 568.5 | 15.6 | vw | |||
| 30 | 595.8 | 62.5 | vw | very broad | ||
| 31 | 602.8 | 8.3 | s | v4(SO4) | O–S–O anti-symmetric bending | |
| 32 | 628.7 | 8.0 | w | |||
| 33 | 658.8 | 8.8 | s | |||
| 34 | 728.4 | 62.2 | vw | very broad | ? | |
| 35 | 752.7 | 29.1 | vw | 772 cm−1 in RRUFF spectrum | ||
| 36 | 799.8 | 47.0 | w | |||
| 37 | 832.0 | 10.9 | vs | v1(SiO4) | Si–O symmetric stretching | |
| 38 | 855.3 | 17.0 | vw | |||
| 39 | 877.8 | 13.3 | s | v3(SiO4) | Si–O anti-symmetric stretching | |
| 40 | 891.0 | 7.2 | w | |||
| 41 | 896.9 | 10.8 | w | |||
| 42 | 940.1 | 17.5 | w | ? | ||
| 43 | 956.9 | 8.7 | w | |||
| 44 | 975.5 | 42.7 | vw | broad | ||
| 45 | 1000.9 | 11.6 | s | hidden | v1(SO4) | S–O symmetric stretching |
| 46 | 1006.0 | 7.7 | vs | |||
| 47 | 1043.9 | 25.0 | w | ? | ||
| 48 | 1120.2 | 13.3 | s | v3(SO4) | S–O anti-symmetric stretching | |
| 49 | 1134.3 | 10.1 | s | |||
| 50 | 1144.0 | 10.0 | w | |||
| 51 | 1181.8 | 8.8 | w | |||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Böhme, N.; Hauke, K.; Neuroth, M.; Geisler, T. In Situ Hyperspectral Raman Imaging of Ternesite Formation and Decomposition at High Temperatures. Minerals 2020, 10, 287. https://doi.org/10.3390/min10030287
Böhme N, Hauke K, Neuroth M, Geisler T. In Situ Hyperspectral Raman Imaging of Ternesite Formation and Decomposition at High Temperatures. Minerals. 2020; 10(3):287. https://doi.org/10.3390/min10030287
Chicago/Turabian StyleBöhme, Nadine, Kerstin Hauke, Manuela Neuroth, and Thorsten Geisler. 2020. "In Situ Hyperspectral Raman Imaging of Ternesite Formation and Decomposition at High Temperatures" Minerals 10, no. 3: 287. https://doi.org/10.3390/min10030287