Rare Earth Element Determination in Uranium Ore Concentrates Using Online and Offline Chromatography Coupled to ICP-MS

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Enriched Stable Isotope Spikes
2.3. Instrumentation
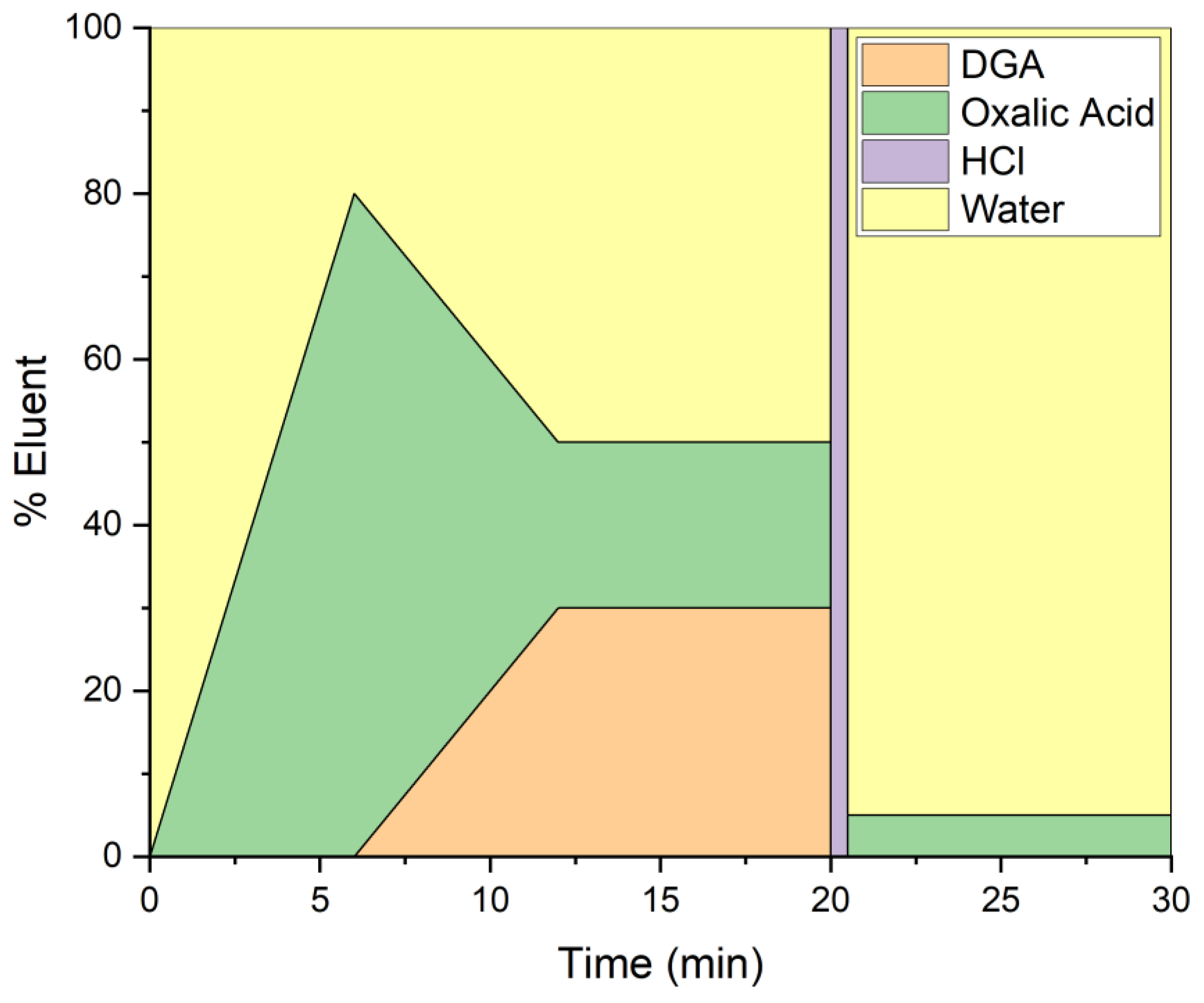
2.4. HPIC Separation
3. Results and Discussion
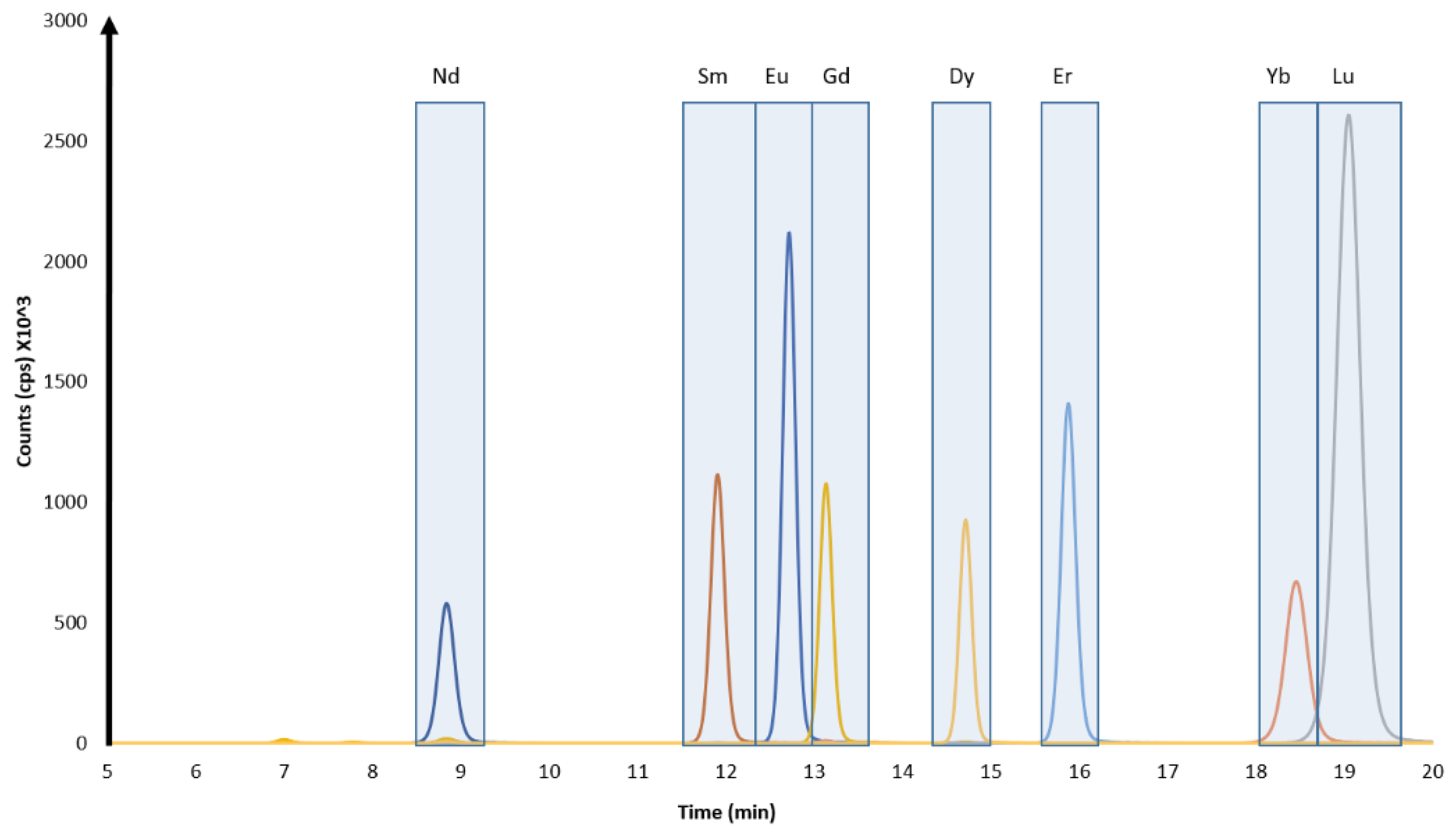
3.1. Chromatographic Separations
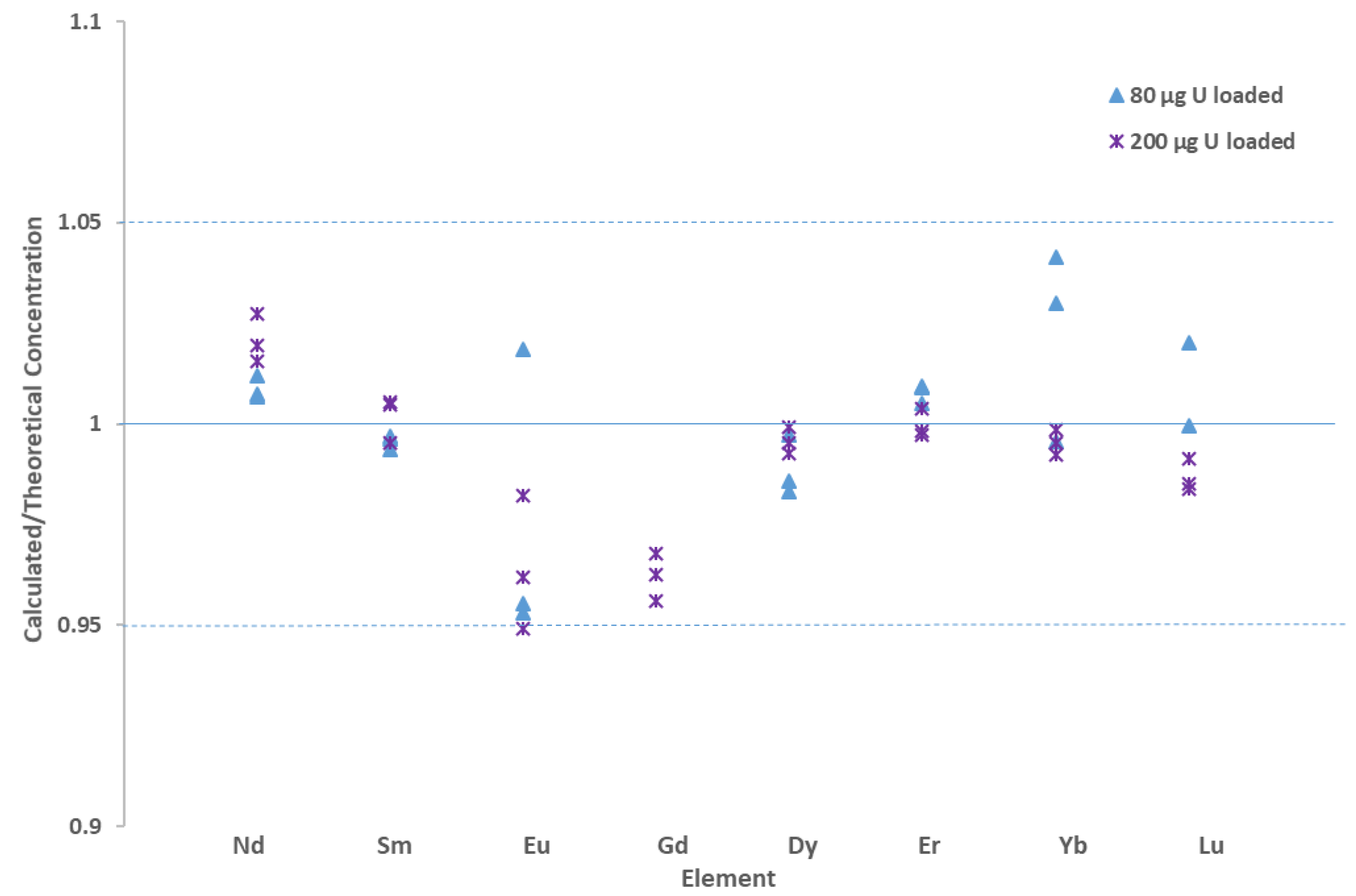
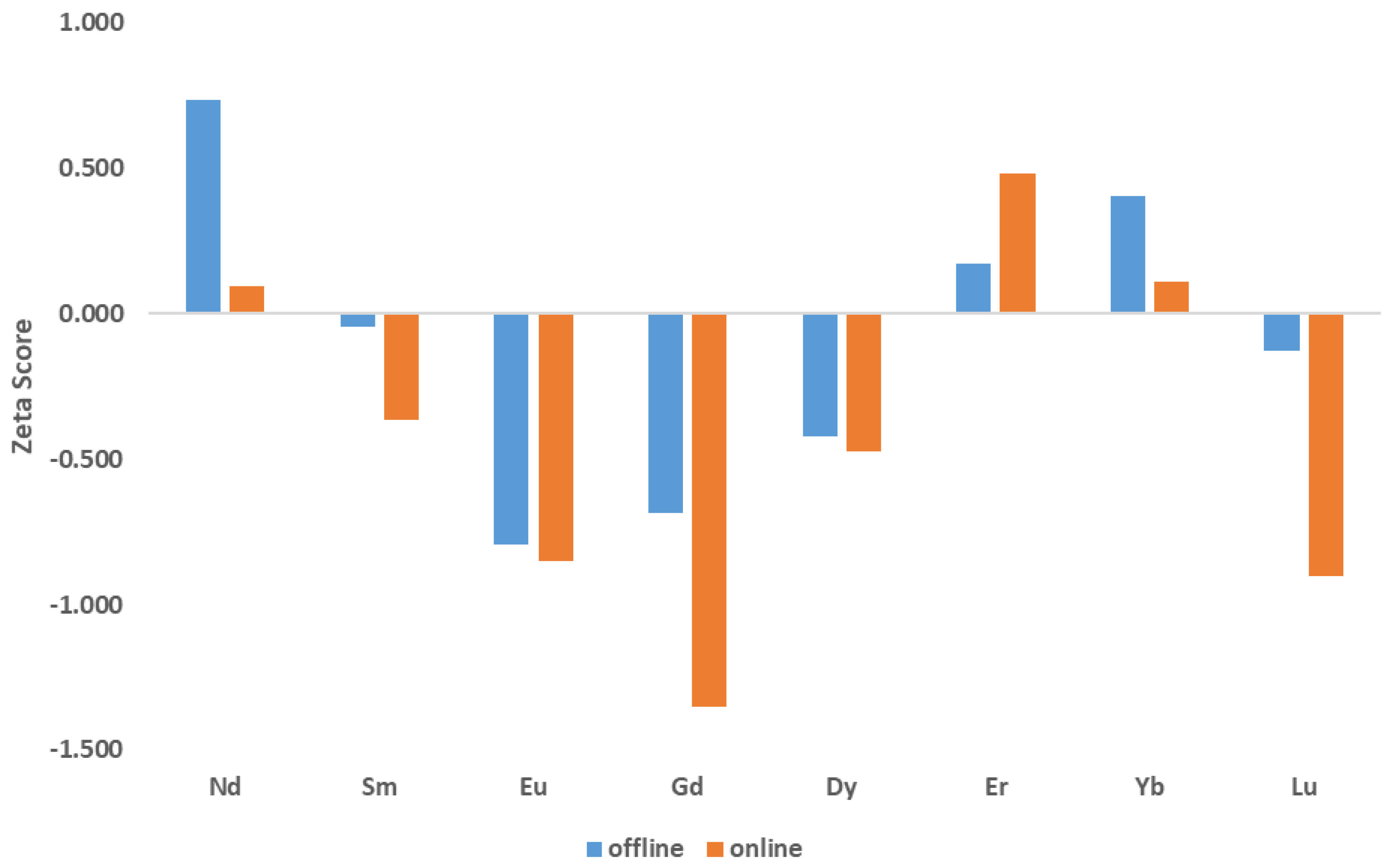
3.2. Offline UOC Separations
3.3. Online UOC Analysis
3.4. Limits of Detection
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Moody, K.J.; Grant, P.M.; Hutcheon, I.D. Nuclear Forensic Analysis; CRC Press: Boca Raton, FL, USA, 2014. [Google Scholar]
- IAEA. World Distribution of Uranium Deposits (UDEPO), with Uranium Deposit Classification, 2009 ed.; International Atomic Energy Agency: Vienna, Austria, 2009. [Google Scholar]
- Keegan, E.; Wallenius, M.; Mayer, K.; Varga, Z.; Rasmussen, G. Attribution of uranium ore concentrates using elemental and anionic data. Appl. Geochem. 2012, 27, 1600–1609. [Google Scholar] [CrossRef]
- Varga, Z.; Katona, R.; Stefánka, Z.; Wallenius, M.; Mayer, K.; Nicholl, A. Determination of rare-earth elements in uranium-bearing materials by inductively coupled plasma mass spectrometry. Talanta 2010, 80, 1744–1749. [Google Scholar] [CrossRef] [PubMed]
- Varga, Z.; Wallenius, M.; Mayer, K.; Meppen, M. Analysis of uranium ore concentrates for origin assessment. Proc. Radiochem. A Suppl. Radiochim. Acta 2011, 1, 27–30. [Google Scholar]
- Varga, Z.; Krajkó, J.; Peńkin, M.; Novák, M.; Eke, Z.; Wallenius, M.; Mayer, K. Identification of uranium signatures relevant for nuclear safeguards and forensics. J. Radioanal. Nucl. Chem. 2017, 312, 639–654. [Google Scholar] [CrossRef] [Green Version]
- Spano, T.L.; Simonetti, A.; Balboni, E.; Dorais, C.; Burns, P.C.; Hixon, A.E. Trace Element Signatures of UOCs: Provenance Indicator. Available online: https://www.researchgate.net/publication/280234351_Trace_Element_Signatures_of_UOCs_Provenance_Indicator (accessed on 20 May 2019).
- Spano, T.L.; Simonetti, A.; Balboni, E.; Dorais, C.; Burns, P.C. Trace element and U isotope analysis of uraninite and ore concentrate: Applications for nuclear forensic investigations. Appl. Geochem. 2017, 84, 277–285. [Google Scholar] [CrossRef]
- Mercadier, J.; Cuney, M.; Lach, P.; Boiron, M.C.; Bonhoure, J.; Richard, A.; Leisen, M.; Kister, P. Origin of uranium deposits revealed by their rare earth element signature. Terra Nova 2011, 23, 264–269. [Google Scholar] [CrossRef]
- Rogers, K.T.; Giaquinto, J.; Essex, R.M.; Metzger, S.C.; Ticknor, B.W.; Hexel, C.R. Trace impurity analysis in uranium oxide via hybrid quantification techniques—gravimetric standard addition and isotope dilution mass spectrometry. J. Radioanal. Nucl. Chem. 2018, 318, 685–694. [Google Scholar] [CrossRef]
- Junior, O.O.; Sarkis, J. Determination of impurities in uranium oxide by inductively coupled plasma mass spectrometry (ICPMS) by the matrix matching method. J. Radioanal. Nucl. Chem. 2002, 254, 519–526. [Google Scholar] [CrossRef]
- Fassett, J. Inorganic Trace Analysis by Isotope Dilution Mass Spectrometry-New. J. Res. Natl. Bur. Stand. 1988, 93, 417–418. [Google Scholar] [CrossRef]
- Fassett, J.D.; Paulsen, P.J. Isotope dilution mass spectrometry for accurate elemental analysis. Anal. Chem. 1989, 61, 643A–649A. [Google Scholar] [CrossRef]
- Perna, L.; Bocci, F.; de las Heras, L.A.; De Pablo, J.; Betti, M. Studies on simultaneous separation and determination of lanthanides and actinides by ion chromatography inductively coupled plasma mass spectrometry combined with isotope dilution mass spectrometry. J. Anal. At. Spectrom. 2002, 17, 1166–1171. [Google Scholar] [CrossRef]
- Vesterlund, A.; Ramebäck, H. Avoiding polyatomic interferences in measurements of lanthanides in uranium material for nuclear forensic purposes. J. Radioanal. Nucl. Chem. 2019, 321, 723–731. [Google Scholar] [CrossRef] [Green Version]
- Olesik, J.W. Elemental analysis using ICP-OES and ICP/MS. Anal. Chem. 1991, 63, 12A–21A. [Google Scholar] [CrossRef]
- Bruzzoniti, M.; Cavalli, S.; Mangia, A.; Mucchino, C.; Sarzanini, C.; Tarasco, E. Ion chromatography with inductively coupled plasma mass spectrometry, a powerful analytical tool for complex matrices: Estimation of Pt and Pd in environmental samples. J. Chromatogr. A 2003, 997, 51–63. [Google Scholar] [CrossRef]
- Wylie, E.M.; Manard, B.T.; Quarles, C.D., Jr.; Meyers, L.A.; Xu, N. An automated micro-separation system for the chromatographic removal of uranium matrix for trace element analysis by ICP-OES. Talanta 2018, 189, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Quarles, C.D., Jr.; Manard, B.T.; Wylie, E.M.; Xu, N. Trace elemental analysis of bulk uranium materials using an inline automated sample preparation technique for ICP-OES. Talanta 2018, 190, 460–465. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Manard, B.T.; Castro, A.; Montoya, D.P.; Xu, N.; Chamberlin, R.M. Solid-phase extraction microfluidic devices for matrix removal in trace element assay of actinide materials. Talanta 2017, 167, 8–13. [Google Scholar] [CrossRef] [Green Version]
- Manard, B.T.; Metzger, S.C.; Quarles, C.D., Jr.; Rogers, K.T.; Ticknor, B.W.; Bostick, D.A.; McBay, E.H.; Hexel, C.R. Evaluation and Specifications for In-Line Uranium Separations Using Inductively Coupled Plasma Optical Emission Spectroscopy (ICP-OES) Detection for Trace Elemental Analysis. Appl. Spectrosc. 2019, 0003702819837650. [Google Scholar] [CrossRef]
- Truscott, J.B.; Jones, P.; Fairman, B.E.; Evans, E.H. Determination of actinides in environmental and biological samples using high-performance chelation ion chromatography coupled to sector-field inductively coupled plasma mass spectrometry. J. Chromatogr. A 2001, 928, 91–98. [Google Scholar] [CrossRef]
- Betti, M.; Menichetti, L.; Moreno, J.B.; Fuoco, R. A preliminary study for the determination of long-lived radionuclides (second transition group 4-d) in environmental samples by ion chromatography inductively coupled plasma-mass spectrometry (IC-ICP-MS). Microchem. J. 2000, 67, 285–290. [Google Scholar] [CrossRef]
- Betti, M. Use of ion chromatography for the determination of fission products and actinides in nuclear applications. J. Chromatogr. A 1997, 789, 369–379. [Google Scholar] [CrossRef]
- Röllin, S.; Kopatjtic, Z.; Wernli, B.; Magyar, B. Determination of lanthanides and actinides in uranium materials by high-performance liquid chromatography with inductively coupled plasma mass spectrometric detection. J. Chromatogr. A 1996, 739, 139–149. [Google Scholar] [CrossRef]
- Schwantes, J.M.; Rundberg, R.S.; Taylor, W.A.; Vieira, D.J. Rapid, high-purity, lanthanide separations using HPLC. J. Alloys Compd. 2006, 418, 189–194. [Google Scholar] [CrossRef]
- Roach, B.D.; Fenske, E.K.; Ilgner, R.H.; Hexel, C.R.; Haverlock, T.J.; Giaquinto, J.M. Development of a fast and efficient analytical technique for the isotopic analysis of fission and actinide elements in environmental matrices. J. Chromatogr. A 2019, 1587, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Fenske, E.K.; Roach, B.D.; Glasgow, D.C.; Ilgner, R.H.; Giaquinto, J.M. Rapid measurements of 235U fission product isotope ratios using an online, high-pressure ion chromatography inductively coupled plasma mass spectrometry protocol with comparison to isotopic depletion models. J. Radioanal. Nucl. Chem. 2019, 320, 153–163. [Google Scholar] [CrossRef]
- Roach, B.D.; Giaquinto, J.M.; Keever, T.J. Integration of a hyphenated HPIC-ICPMS protocol for the measurement of transplutonium isotopic mass distributions for 252Cf campaigns at Oak Ridge National Laboratory. J. Radioanal. Nucl. Chem. 2018, 318, 407–413. [Google Scholar] [CrossRef]
- Cotton, S. Lanthanide and Actinide Chemistry; John Wiley & Sons: Hoboken, NJ, USA, 2013. [Google Scholar]
- Nash, K.L. A review of the basic chemistry and recent developments in trivalent f-elements separations. Solvent Extr. Ion Exch. 1993, 11, 729–768. [Google Scholar] [CrossRef]
- Eppich, G.; Kips, R.; Lindvall, R. Compilation of LLNL CUP-2 Data; Technical Report; Lawrence Livermore National Lab.: Livermore, CA, USA, 2016. [Google Scholar] [CrossRef]
- Roach, B.D.; Fenske, E.K.; Glasgow, D.C.; Partridge, J.D.; Keever, T.J.; Giaquinto, J.M. Rapid concentration and isotopic measurements of ultra-trace 235U fission products with comparison to an ORIGEN isotope depletion model. Talanta 2019. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Element | Isotope | Natural Abundance % | Isobaric Interference (Natural Abundance %) | Polyatomic Interferences |
|---|---|---|---|---|
| Nd | 146 | Nd (17.19) | 130Ba16O, 98Ru16O3 | |
| 148 * | Nd (5.76) | 132Ba16O, 100Ru16O3 | ||
| Sm | 150 * | Sm (7.38) | Nd (5.64) | 134Ba16O, 102Ru16O3 |
| 152 | Sm (26.75) | Gd (0.2) | 136Ba16O, 136Ce16O | |
| Eu | 151 * | Eu (47.81) | 135Ba16O | |
| 153 | Eu (52.19) | 137Ba16O | ||
| Gd | 152 * | Gd (0.2) | Sm (26.75) | 136Ba16O, 136Ce16O |
| 154 | Gd (2.18) | Sm (22.75) | 138Ba16O, 138La16O | |
| Dy | 160 * | Dy (2.33) | 144Nd16O, 144Sm16O | |
| 161 | Dy (18.90) | 145Nd16O | ||
| Er | 164 * | Er (1.60) | Dy (28.26) | 148Nd16O |
| 166 | Er (33.50) | 150Sm16O, 150Nd16O | ||
| Yb | 173 * | Yb (16.10) | 157Gd16O | |
| 174 | Yb (32.03) | 158Gd16O | ||
| Lu | 175 | Lu (97.40) | 159Gd16O, 159Tb16O | |
| 176 * | Lu (2.60) | Yb (13.00) | 160Dy16O |
| Element | Reported Concentration (µg g−1 U) | Offline Concentration (µg g−1 U) | Difference % | Online Concentration (µg g−1 U) | Difference % |
|---|---|---|---|---|---|
| Nd | 26.29 ± 0.38 | 26.68 ± 0.37 | +1.47 | 26.37 ± 1.07 | +0.31 |
| Sm | 12.29 ± 0.13 | 12.28 ± 0.18 | −0.13 | 12.17 ± 0.30 | −1.02 |
| Eu | 1.01 ± 0.02 | 0.98 ± 0.04 | −3.00 | 0.96 ± 0.06 | −5.36 |
| Gd | 19.48 ± 0.28 | 19.16 ± 0.37 | −1.68 | 18.50 ± 0.67 | −5.0 |
| Dy | 24.72 ± 0.38 | 24.52 ± 0.32 | −0.78 | 24.45 ± 0.42 | −1.08 |
| Er | 11.50 ± 0.20 | 11.54 ± 0.12 | +0.39 | 11.69 ± 0.34 | +1.71 |
| Yb | 9.05 ± 0.10 | 9.13 ± 0.10 | +0.88 | 9.08 ± 0.28 | +0.31 |
| Lu | 1.09 ± 0.02 | 1.09 ± 0.02 | −0.12 | 1.05 ± 0.05 | −3.98 |
| Element | Offline LOD (µg g−1 U) | Offline LOQ (µg g−1 U) | Online LOD (µg g−1 U) | Online LOQ (µg g−1 U) |
|---|---|---|---|---|
| Nd | 0.044 | 0.15 | 0.0084 | 0.028 |
| Sm | 0.0095 | 0.032 | 0.018 | 0.059 |
| Eu | 0.028 | 0.094 | 0.022 | 0.072 |
| Gd | 0.34 | 1.14 | 0.0093 | 0.01 |
| Dy | 0.021 | 0.069 | 0.0017 | 0.0055 |
| Er | 0.0085 | 0.028 | 0.0038 | 0.013 |
| Yb | 0.0052 | 0.017 | 0.00091 | 0.0031 |
| Lu | 0.0075 | 0.025 | 0.0015 | 0.0051 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bradley, V.C.; Manard, B.T.; Roach, B.D.; Metzger, S.C.; Rogers, K.T.; Ticknor, B.W.; Wysor, S.K.; Brockman, J.D.; Hexel, C.R. Rare Earth Element Determination in Uranium Ore Concentrates Using Online and Offline Chromatography Coupled to ICP-MS. Minerals 2020, 10, 55. https://doi.org/10.3390/min10010055
Bradley VC, Manard BT, Roach BD, Metzger SC, Rogers KT, Ticknor BW, Wysor SK, Brockman JD, Hexel CR. Rare Earth Element Determination in Uranium Ore Concentrates Using Online and Offline Chromatography Coupled to ICP-MS. Minerals. 2020; 10(1):55. https://doi.org/10.3390/min10010055
Chicago/Turabian StyleBradley, Veronica C., Benjamin T. Manard, Benjamin D. Roach, Shalina C. Metzger, Kayron T. Rogers, Brian W. Ticknor, Sarah K. Wysor, John D. Brockman, and Cole R. Hexel. 2020. "Rare Earth Element Determination in Uranium Ore Concentrates Using Online and Offline Chromatography Coupled to ICP-MS" Minerals 10, no. 1: 55. https://doi.org/10.3390/min10010055