1. Introduction
The development of multifunctional nanoparticles has a huge impact on the future of personalized medicine. Nanoparticles can be used for therapeutic purposes in anti-cancer therapy at the molecular level, which has been difficult so far. In earlier work [
1,
2,
3], an enzyme GOx (3QVR) that fulfills the role of a biosensor, had been used for immobilization on the gel, while the polymer was polyethylenimine (PEI). By assembling polymeric nano-gels (for example PEI) and antibodies on nano-molecules [
1,
2,
3], it was possible to recognize receptors of certain integrins on lung cancer tissues and to identify new cancer vessels.
In the present article, polyethylenimines (PEI) were studied. Molecular docking analysis of fifteen PEI derivatives acting as ligands on some cube rhombellane homeomorphs was carried out for the first time. Fourteen types of cube rhombellanes and three groups of polyethylenimines (PEIs), namely, branched (B-PEI), linear (L-PEI) and dendrimer (D-PEI) were used.
The choice of ligands and docked nanostructures was guided by our earlier studies [
1,
2,
3,
4,
5,
6,
7,
8].
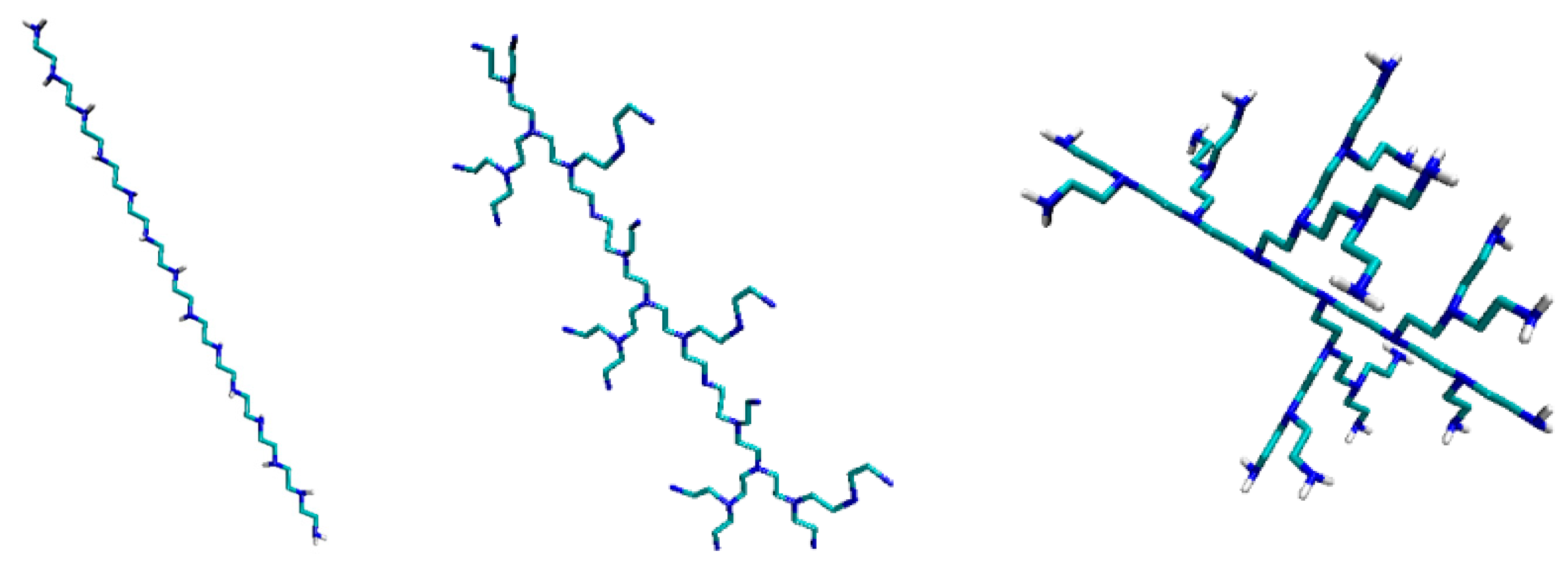
PEIs (polyethylenimines) are polymeric molecules built of two aliphatic carbons and repeating units of amine groups. There are L-, B-, and DPEI (
Figure 1). Linear PEI (LPEI) are built of secondary and primary amino groups (
Figure 1, left); branched PEI (B-PEI) are built of all types amino groups such as primary, secondary and tertiary (
Figure 1, middle), while dendrimers [
9,
10] are symmetric around the core (
Figure 1, right). PEI despite the fact that are cytotoxic [
11], have many applications, first of all, as transfection reagents [
12].
Calculations at the B3LYP/6-31G (d, p) level of theory [
16,
17,
18,
19] confirmed the hypothesis that rhombellanes are energetically feasible in the hope of a real synthesis [
20,
21,
22,
23,
24,
25,
26].
Rhombellanes have certain specific traits which define these group of structures. At first, all strong rings are squares/rhombs. The second vertex classes consist of only non-connected vertices. Omega polynomial has a single term: 1X^|E| and they contain one K2.3 complete bipartite subgraph or the smallest rhombellane rbl.5. The end line graph of the parent graph has a Hamiltonian circuit.
To explore the internal molecular mobility that is important in the bioactivity study of these compounds we used the Molecular mechanics (MMFF94) [
27] method. In this way we study the pharmaceutical important parameters of rhombellanes [
27].
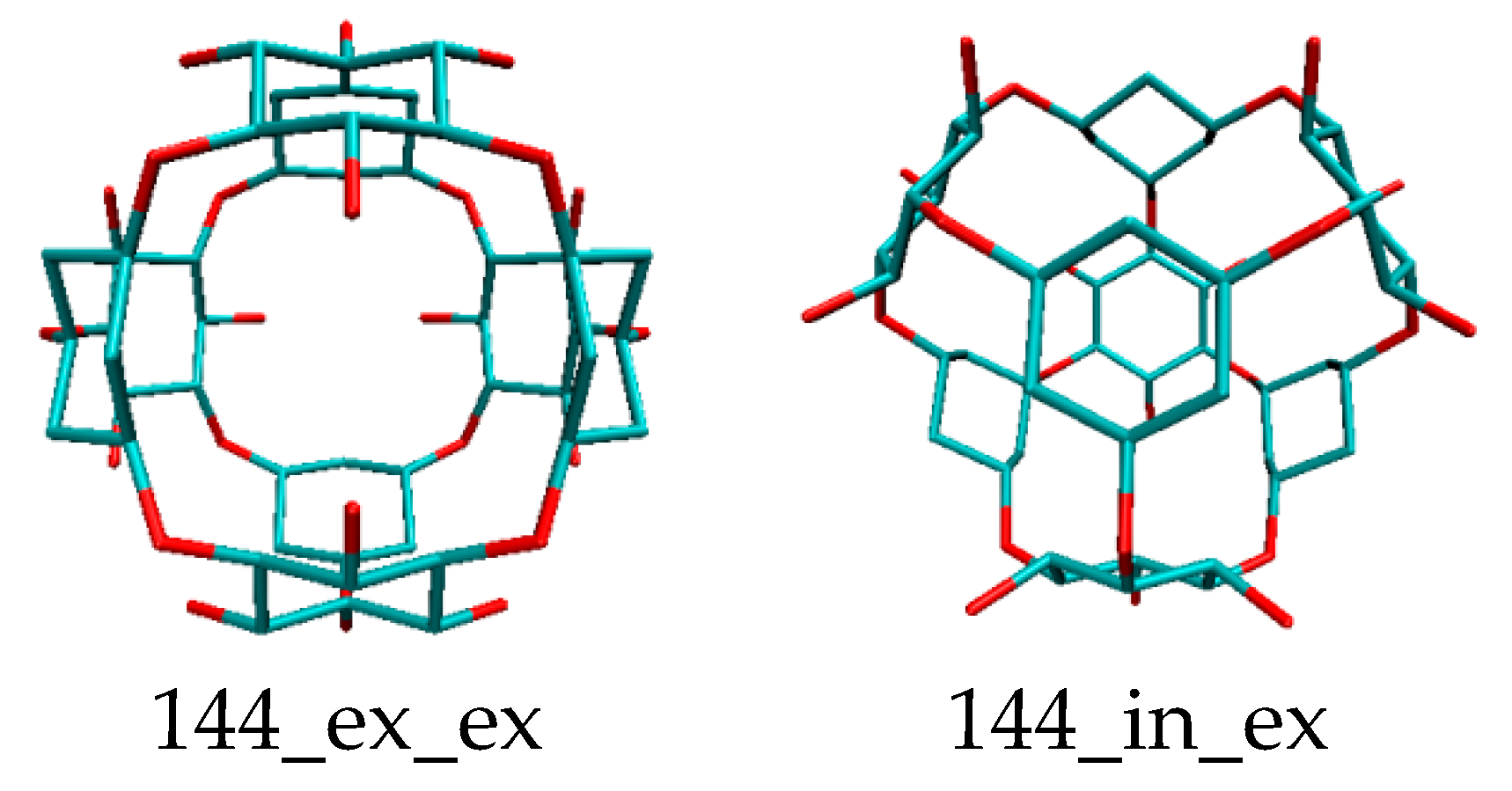
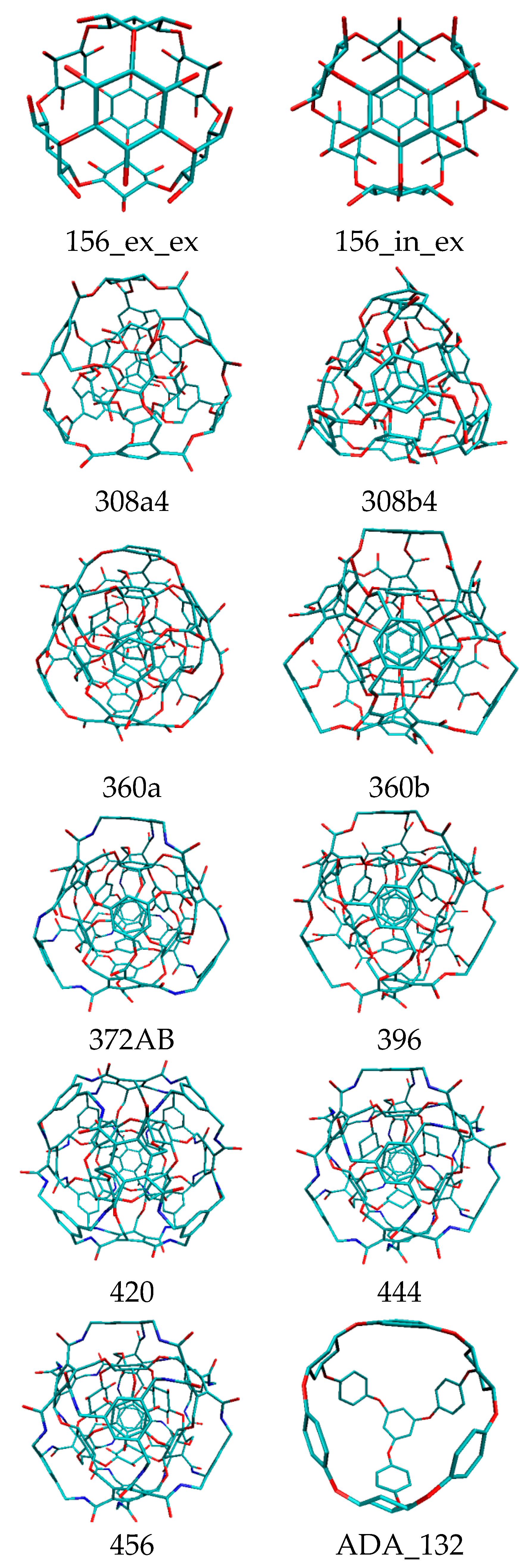
Rhombellans (
Figure 2) seem to be structures suitable for medical chemistry, with a new class of structures which could be an used in personalized medicine as new carrier nanostructures.
Because rhombellane homeomorphs may be bound to a protein, an attempt was made to deposit PEI derivatives on rhombellanes, as possible nano-drug complexes. Detailed analysis of structural properties after docking showed many interesting features. Behavior of polyethylenimine (linear LPEI, branched BPEI and/or dendrimers DPEI) with respect to rhombellane homeomorphs, in terms of their (interacting) topology, geometry and energy, was studied. After the docking procedure, the best values of ligand–rhombellane affinity were found, which is an important result for homeomorphs.
The article is a collection of new data in the new field of rhombellanes (
Figure 2).
3. Results and Discussion
The results are presented in the following tables and figures. Rhombellane structures are given by their atom number.
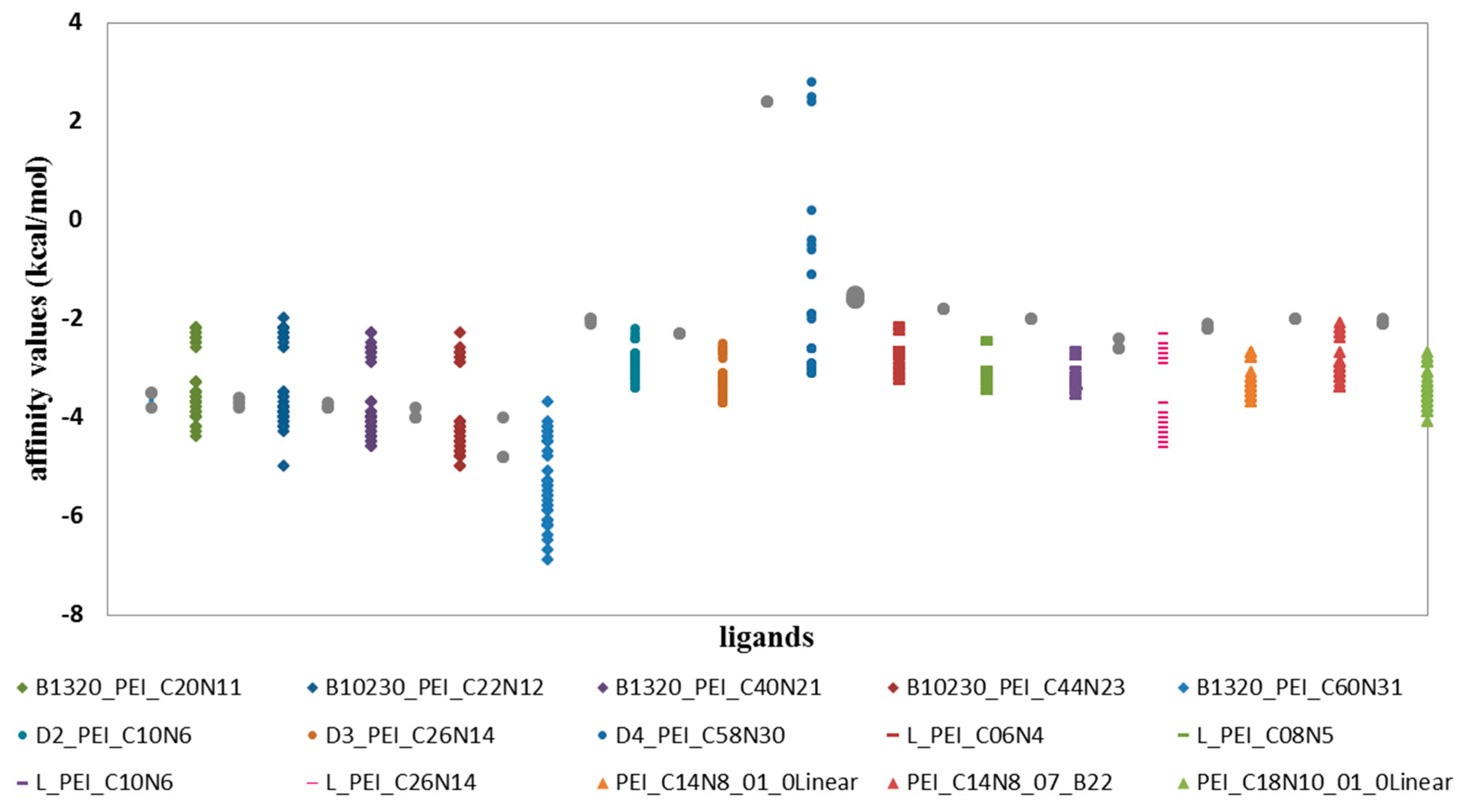
In relation to the ligands of the D and L groups, the largest affinity values of the ligand–fullerenes were found for all ligands from B group (
Figure 3,
Table 1), for which the affinity values range from −2 to −7 kcal/mol. With the B_1320_PEI_C60N31 ligand the values are the lowest, thus showing the best affinity for all proposed fullerenes, with affinity values from −4 to −7 kcal/mol. All values of the interactions were compared with the values for C
60 fullerene, which was, as always, used as the reference structure in nanostructures family. Therefore, in all cases of ligands from the B group, there are affinities with better and worse values of energy compared with affinity ligand–fulleren C
60 (
Figure 3,
Table 1).
When comparing the values of affinity of D ligand–fulleren and L ligand–fulleren complexes, it is clear that these complexes show the lowest values with all study fullerenes, compared with fulleren C
60 (
Figure 3,
Table 2 and
Table 3).
For the other proposed ligands of type D, L and PEI_C14N8_01_Linear; PEI_C14N8_07_B22; PEI_C18N10_01_0, their affinities ranged from −2 to −4 kcal/mol (
Table 2 and
Table 3). However, the ligand D4_PEI_C58N30 shows not only affinity in this range, i.e., (−2 to −4 kcal/mol), but also low affinities for fullerenes with high values of around 0 kcal/mol, and it does not even have the possibility of interacting with fullerenes as their affinity values are positive (
Figure 3,
Table 2 and
Table 3)
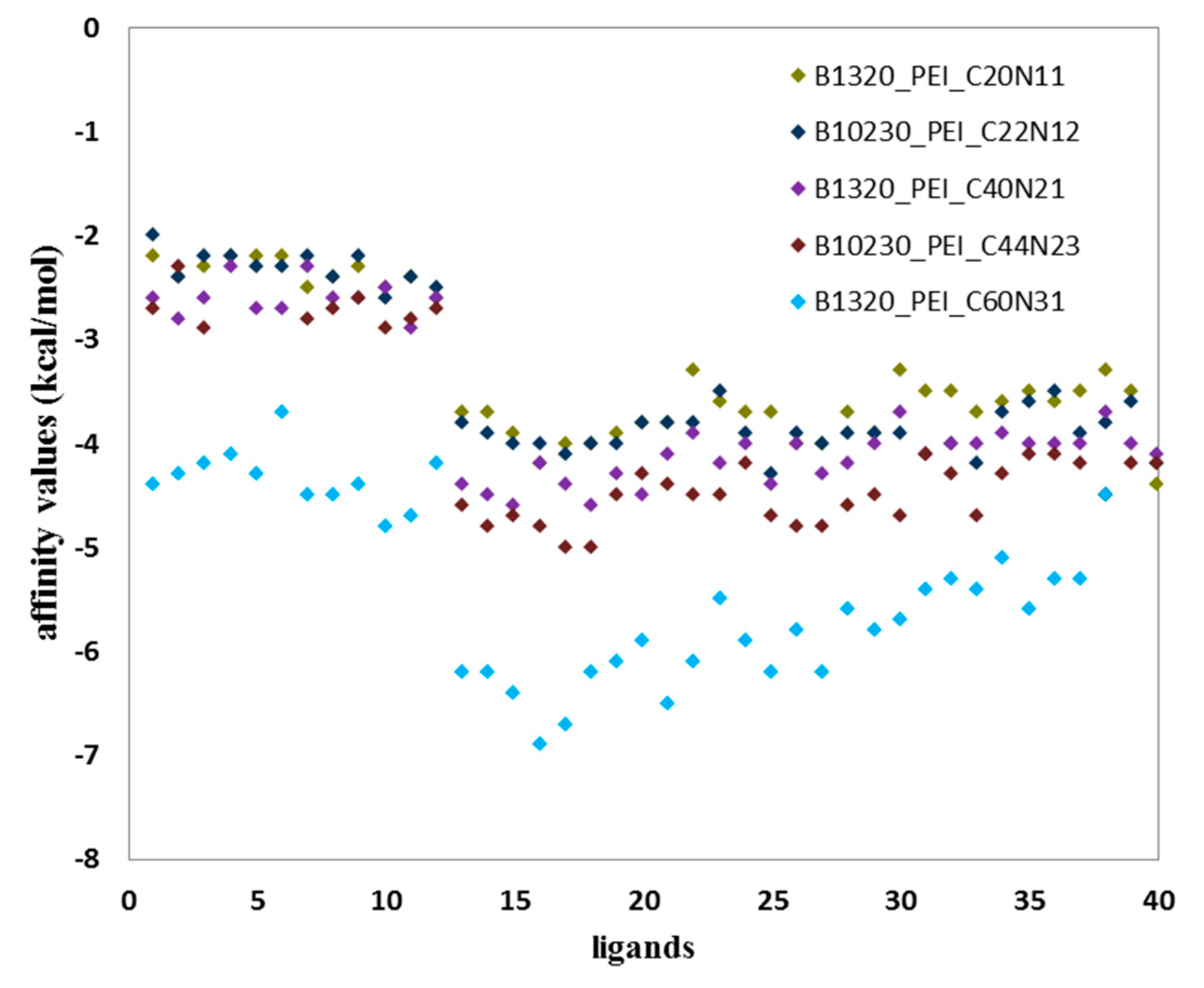
The diagram (
Figure 4) shows two populations of affinity values of ligand B–fullerenes. The first with values ranging from −2 to −3 kcal/mol and in the case of second population from −3.3 to −5 kcal/mol for B1320_PEI_C20N11; B10230_PEI_C22N12; B1320_PEI_C40N21; B10230_PEI_C44N23.
Similarly, two populations are visible in the case of B1320_PEI_C60N31 with markedly reduced affinity values relative to the values of the affinity represented by the first four ligands of the B group, described above (
Table 3 and
Table 4). The first population has interaction values ranging from −4 kcal/mol to −5 kcal/mol, the second shows much higher affinity values from −5 to −7 kcal/mol (
Table 3 and
Table 4).
Two populations are also visible for other ligands from the D and L groups forming interactions with tested fullerenes. Quantitatively, the largest number of ligand–fullerene interactions is expressed by affinity values in the range of −3 to −4 kcal/mol, and this is a representative population. The second population is expressed by a small representation of the number of affinities with values ranging from −2 to −3 kcal/mol.
The existence of these two populations is closely related to the interaction with two fullerene groups. Small structures of ligands easily react with small structures of nanoparticles and vice versa.
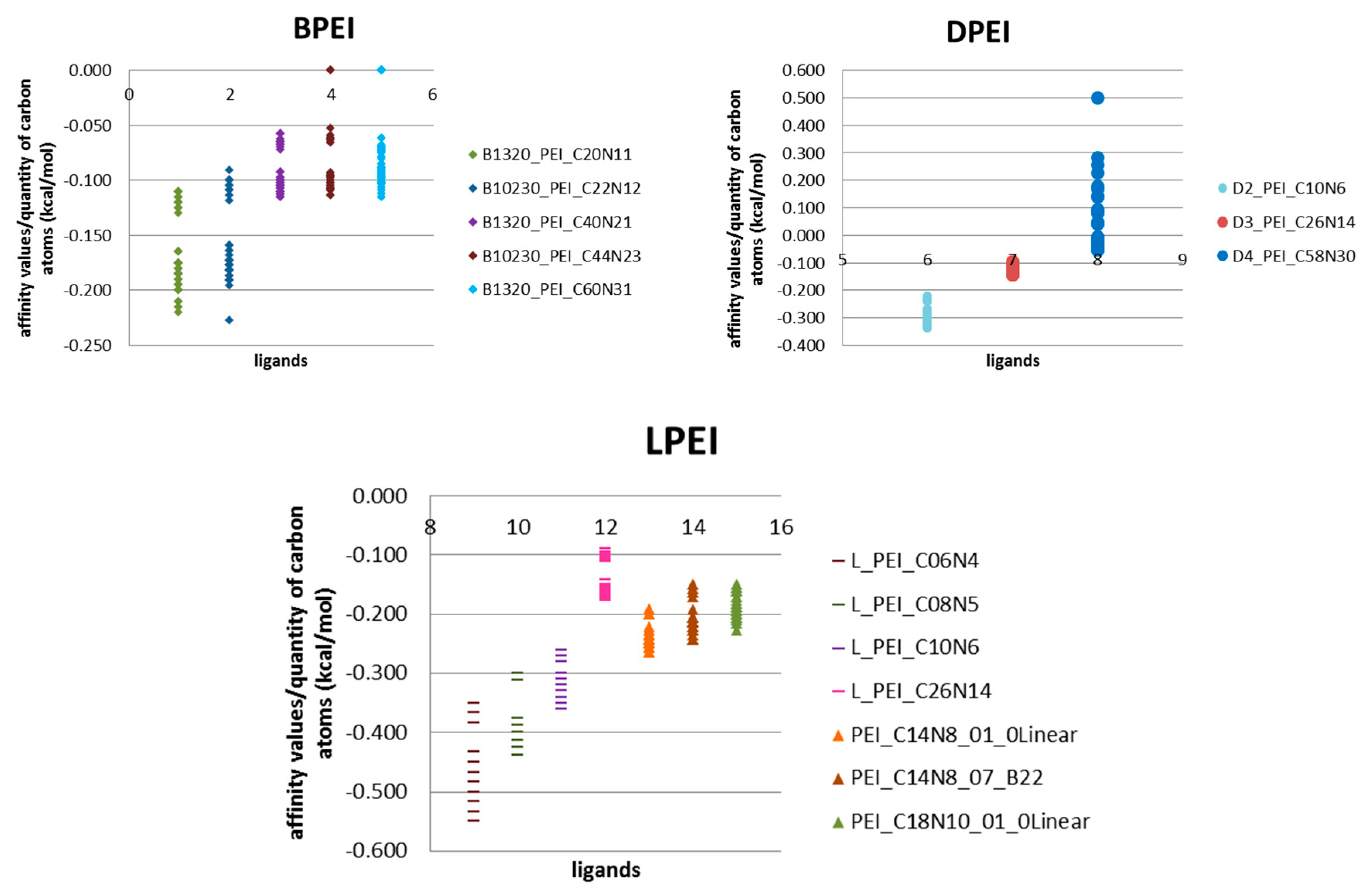
Unfortunately, the energy parameter itself is insufficient. By using energy per quantity of carbon atoms (kcal/mol) parameter it is clearly visible that the elongation of carbon chain does not affect the binding efficiency, but only increases affinity (
Figure 5).
The best binding efficiency is shown by linear ligands L, with highest values of this parameter, compared with values of ligands from B and D groups (
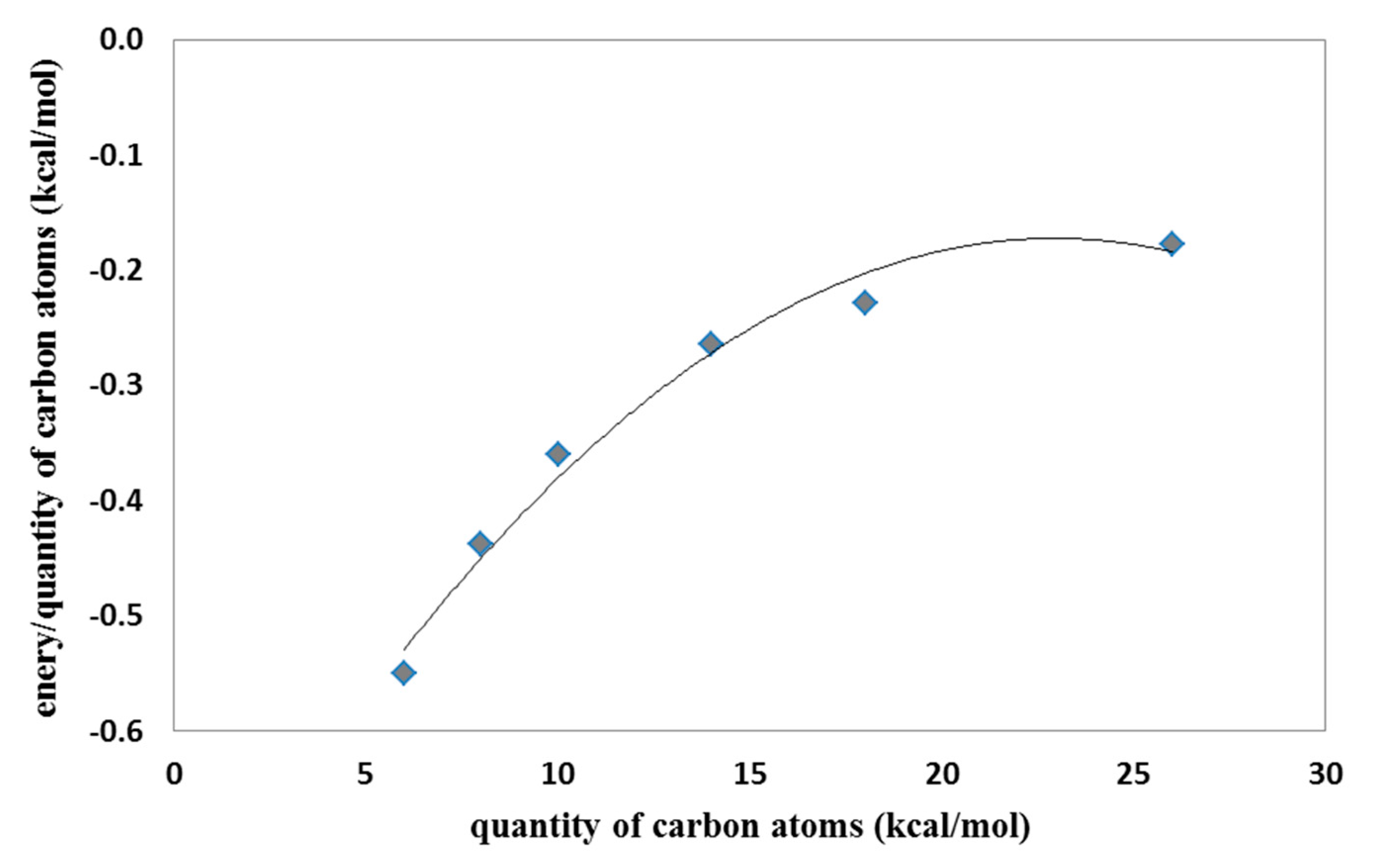
Figure 5). The shorter the molecule, the better the binding performance, the more the particle grows and the lower the yield. For linear LPEI structures, as the carbon chain length increases, the binding efficiency decreases and the saturation around the length of the chain with twenty carbon atoms is clearly visible (
Figure 6).
Similar observations have been made for group B ligands. Twofold chain elongation results in a two-fold decrease in binding efficiency (
Figure 5,
Table 1). In the case of dendrimeric structures, as the complexity of the system increases, the value of binding efficiency decreases (
Figure 5,
Table 2). Among the fullerenes tested, the best effects were found for fullerenes ADA and 308a4/b4. Small ligands easily form complexes primarily with fullerene ADA, long-chain ligands interact with the 308a4/b4 nanostructure (
Figure 5). Thus, for small ligands, the best binding efficiency is with small fullerenes, while large fullerenes require large ligands.
The best binding affinity of ligands BPEI (
Table 4) DPEI (
Table 5) and LPEI (
Table 6 and
Table 7), k max values of binding constant estimated with use of binding free energy obtained for the best complex of ligand with nanostructure and k max differences relative C
60 molecule, defining the difference in the quality of ligand binding with considered nanostructure in comparison to reference system, were estimated for the tested Rbl-structures in relation to the affinity value obtained for the fullerene C
60. The highest positive percentage deviations from the affinity of ligands to fullerene C
60 were obtained for those Rbl-structures showing the highest binding values (
Table 4,
Table 5,
Table 6 and
Table 7, in boldface). Two last columns show the equilibrium K value of the bonds.
The higher the K value, the more the reaction proceeds towards the formation of the complex.
Detailed analysis of structural properties after docking showed that the affinities of the ligands to the rhombellanes surface are correlated with the quality of hydrogen bonds formed between them. The distance between acceptor and hydrogen atoms is the criterion for classification of the strength of hydrogen bonds: weak interactions are characterized by distances <3 Å, strong interactions by a distance <1.6 Å and medium strength by values in the range from 1.6 Å to 2.0 Å.
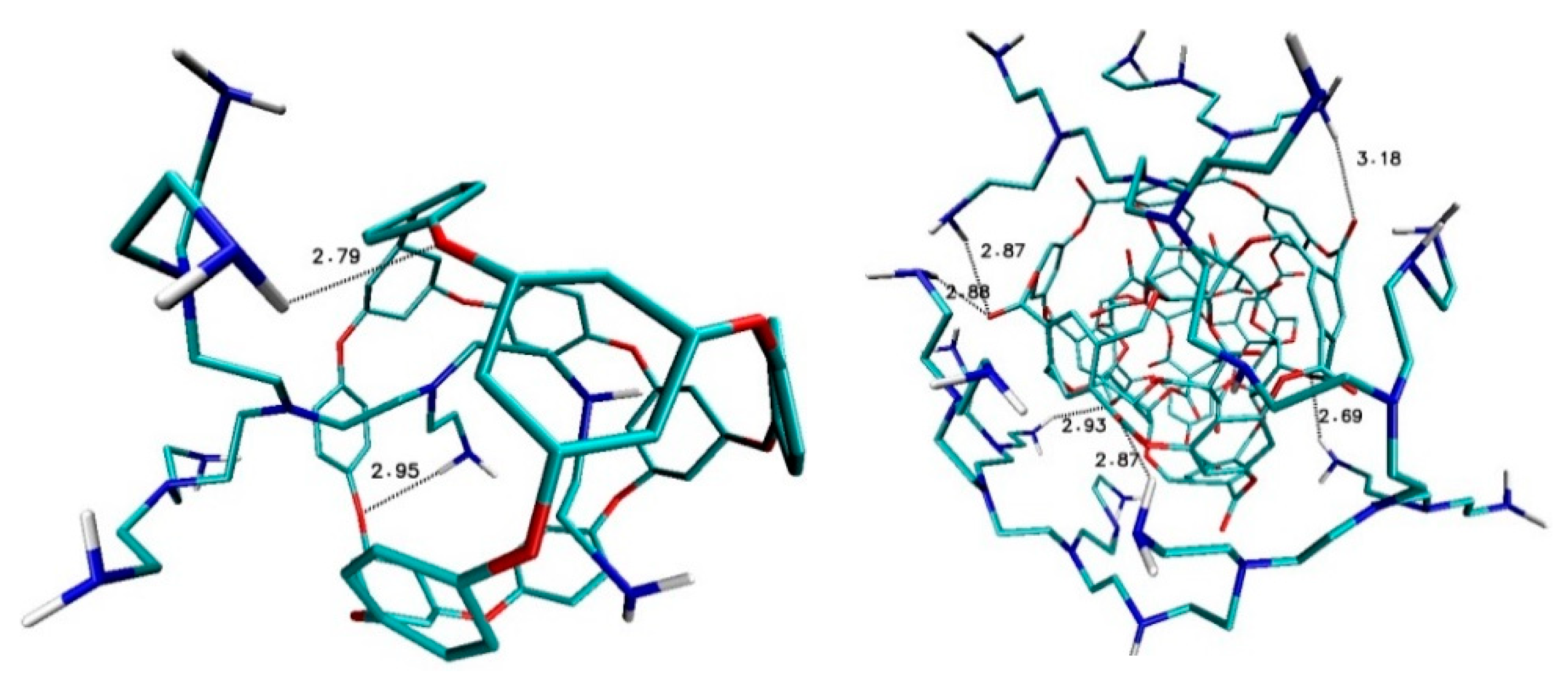
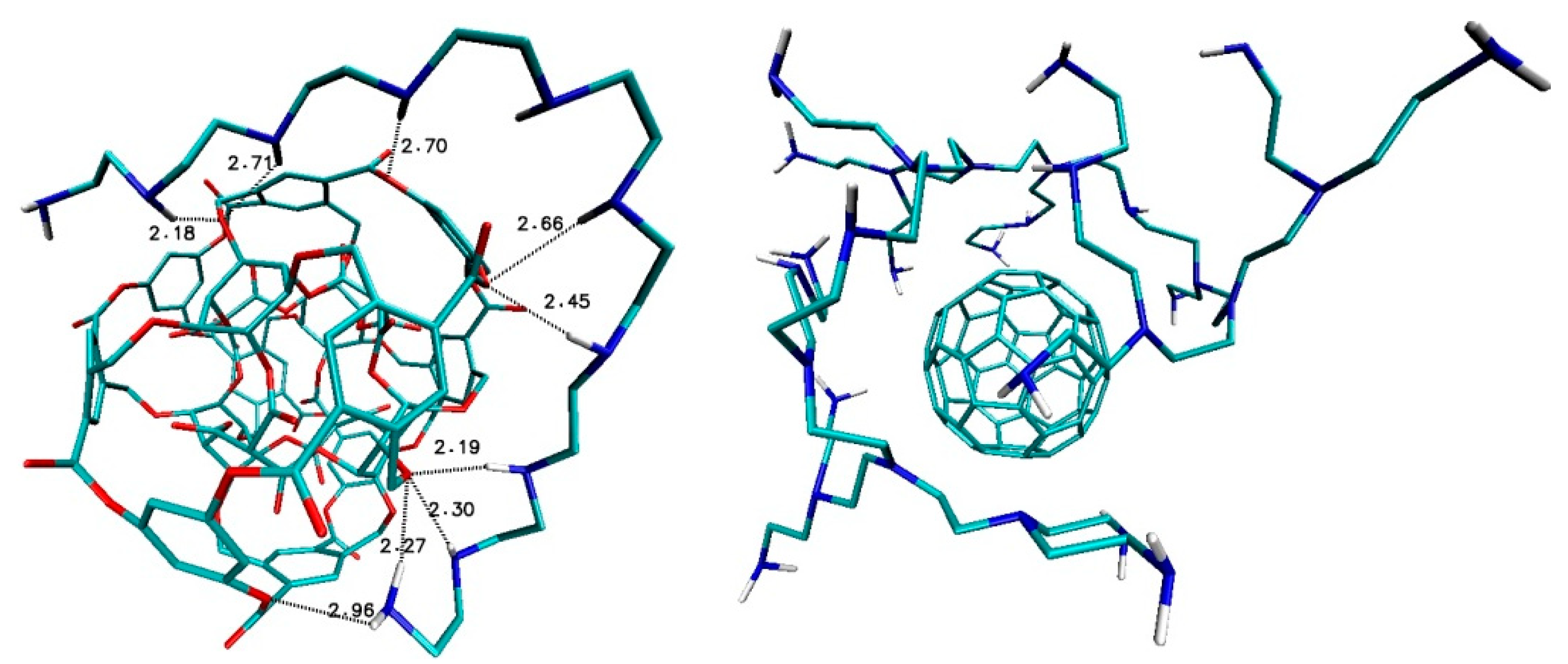
Ligand B1320_PEI_C20N11 and nanostructure ADA_132 form two hydrogen bonds with medium strength between amino groups of ligand and oxygen atoms of fullerenes with binding lengths 2.79Å and 2.95Å (
Figure 7). Also, in the case of ligand B1320_PEI_C60N31–fullerene 308b4 four medium hydrogen bonds were created with bond lengths 2.69 Å, 2.87 Å and 2.93 Å (
Figure 7).
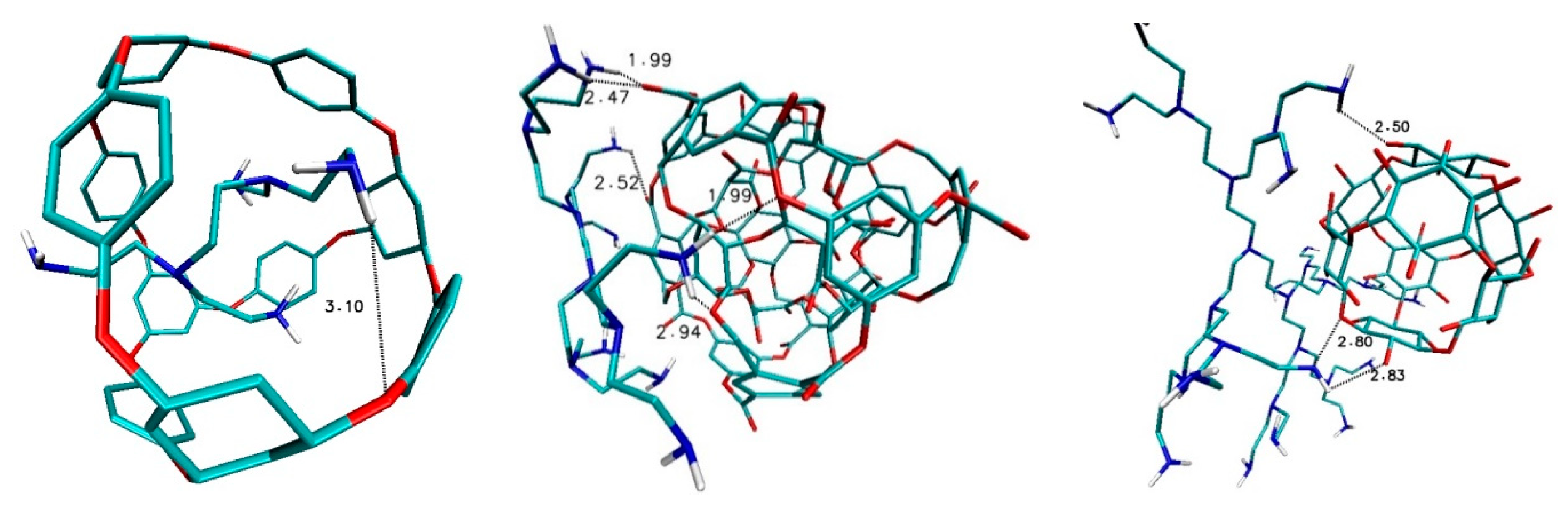
After the docking of ligands from D group, different interactions could be found, namely in the case of fullerene ADA_132 and ligands D2_PEI_C10N6 there is only one week hydrogen bond, while for fullerene 308b4 with D3_PEI_C26N14 ligand there are several strong and medium hydrogen bonds, first of all between amino groups of ligand and oxygen atom of nanostructure with bond lengths 1.9 Å, 2.47 Å., 2.52 Å and 2.94 Å. In the case of fullerene 156_in_ex with ligand D4_PEI_C58N30, there are three hydrogen bonds of medium strength; with bond lengths 2.50 Å, 2.80 Å and 2.83 Å (
Figure 8).
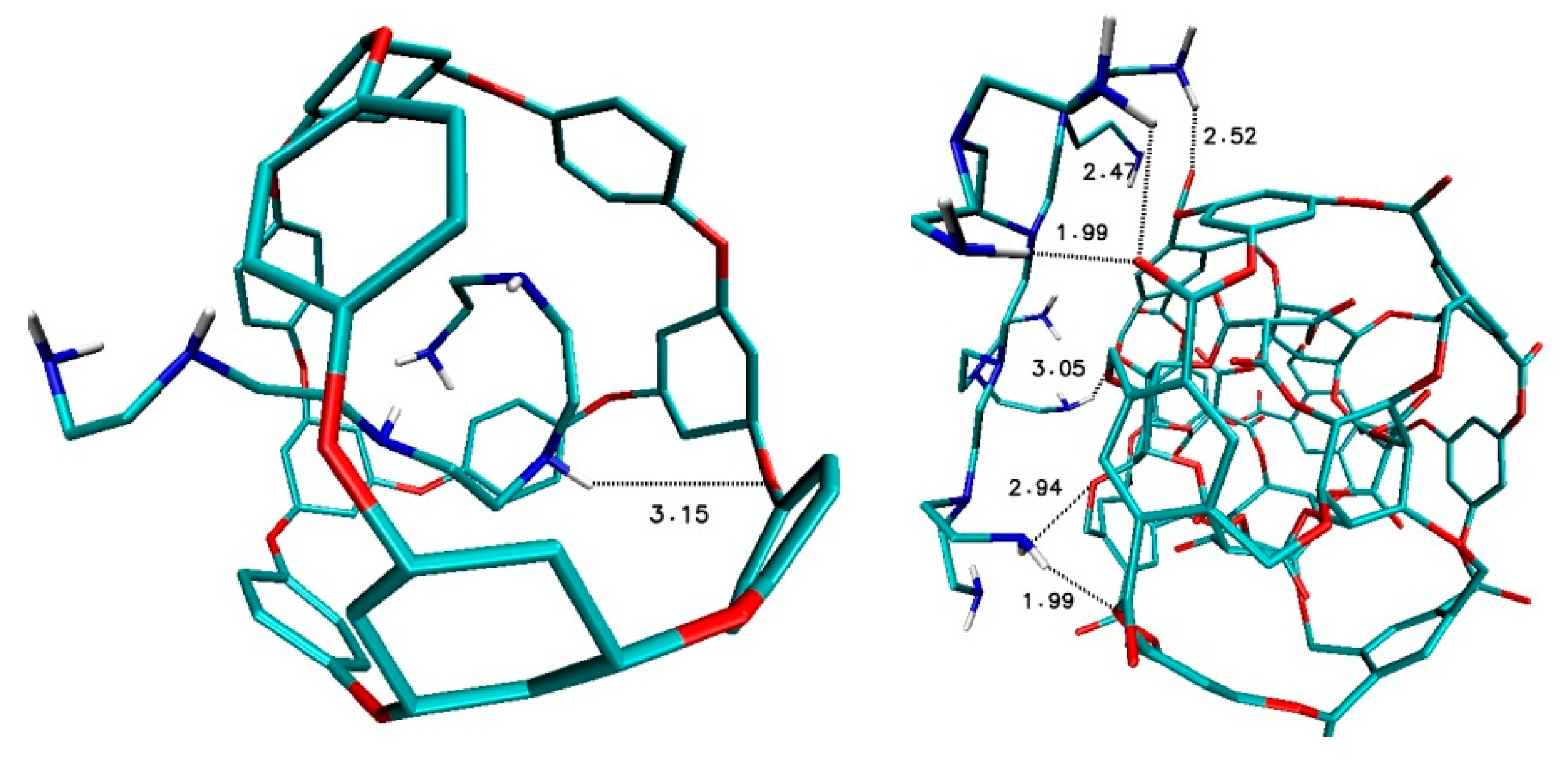
After the docking of ligands from L group, different interactions were also found, namely in the case of fullerene ADA_132 and ligands L_PEI_C10N6 there is only one-week hydrogen bond, the same as in the case D2_PEI_C10N6-ADA_132 (
Figure 9). Again, as in DPEI-308b4 case, there are many interactions between hydrogen atoms of nitrogen groups of ligand and oxygen atoms of nanostructure with values 1.99 Å, 2.47 Å, 2.52 Å, 2.94 Å and 3.05 Å (
Figure 9).
In the case of fullerene 308b4 and ligand PEI_C18N10_01_0Linear, there are several strong and medium hydrogen bonds, while in the case of fullerene C
60 as references structure with B1320_PEI_C60N31 ligand there are no important interactions (
Figure 10).
4. Conclusions
As a proposal for a new nanodrug, an attempt was made to implement PEI ligands on the cube rhombellane homeomorphic surface. Fourteen types of cube rhombellanes were used together with three groups of polyethylenimines (PEIs), namely, branched (B-PEI), linear (L-PEI) and dendrimer (D-PEI). Ligand-fullerenes interactions were described in terms of quality and quantity. Specifically, there were calculated the affinity values and affinity per quantity of carbon atoms after the docking procedure for ligand nanostructure. The best binding efficiency was shown by linear ligands L, with highest values of this parameter, compared with values of ligand from B and D groups. The shorter the molecule, the better the binding performance, the more the particle grows and the lower the yield. For linear structures LPEI, as the carbon chain length increases, the binding efficiency decreases and the saturation around the length of the chain with twenty carbon atoms is clearly visible. Similar observations have been made for group B ligands. Twofold chain elongation results in a two-fold decrease in binding efficiency. In the case of dendrimeric structures, as the complexity of the system increases, the value of binding efficiency decreases. Two populations of affinity values have been observed, which is closely related to the interaction with two fullerene groups. Small structures of ligands easily react with small structures of nanoparticles and vice versa. The best binding affinity of ligands and k max values of binding constant were estimated with the use of binding free energy obtained for the best complex of ligand with nanostructure. Also, k max differences relative C60 molecule, defining the difference in quality of ligand binding with considered nanostructure in comparison to reference system, were calculated. The highest positive percentage deviations were obtained for ligand–fullerene complexes showing the highest binding energy values. Detailed analysis of structural properties after docking showed that the values of affinity of the studied indolizine ligands to the Rhombellanes surface are correlated with the strength/length of hydrogen bonds formed between them.














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}