
Immunotherapy Strategy for Systemic Autoimmune Diseases: Betting on CAR-T Cells and Antibodies
 ,
,  , , and
, , and 
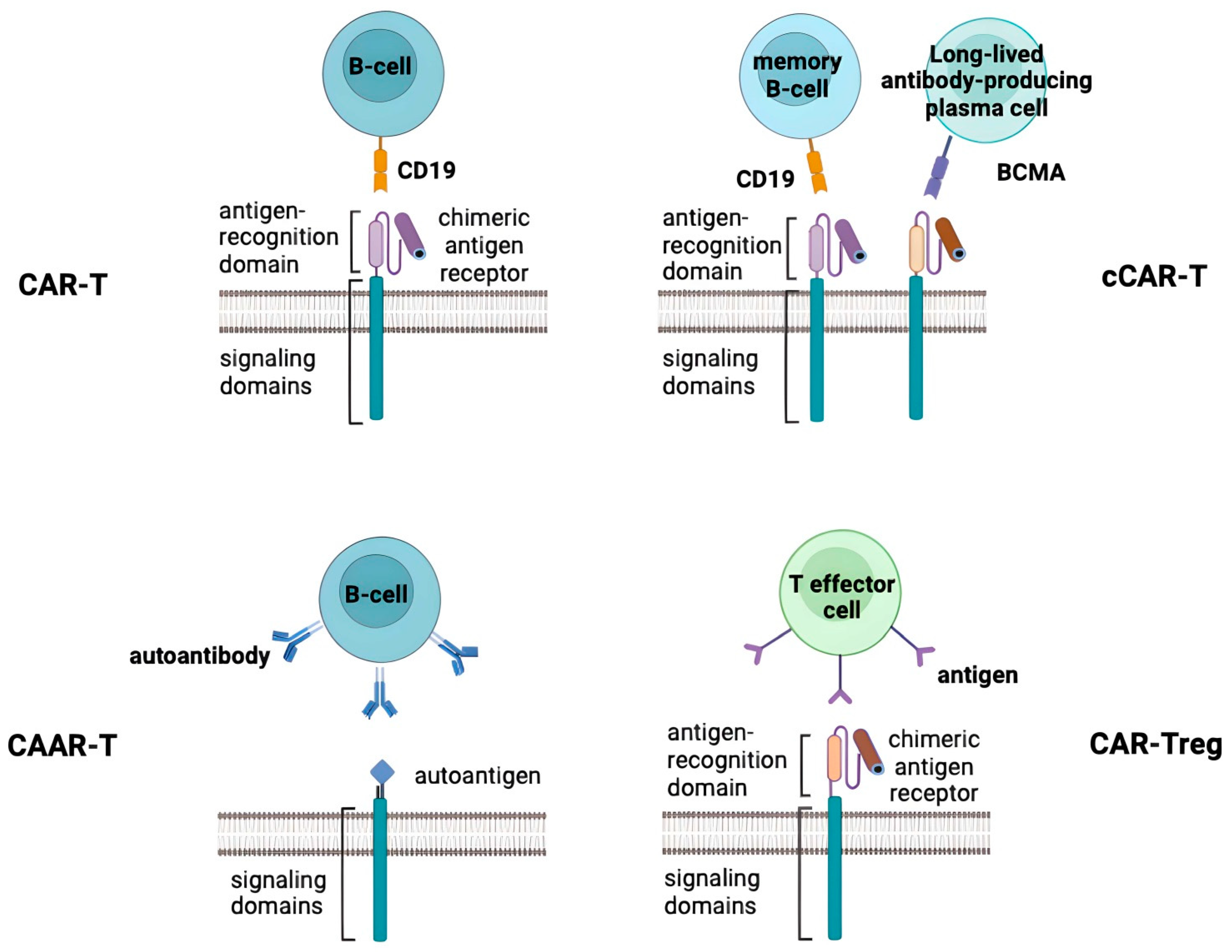{kind=link}
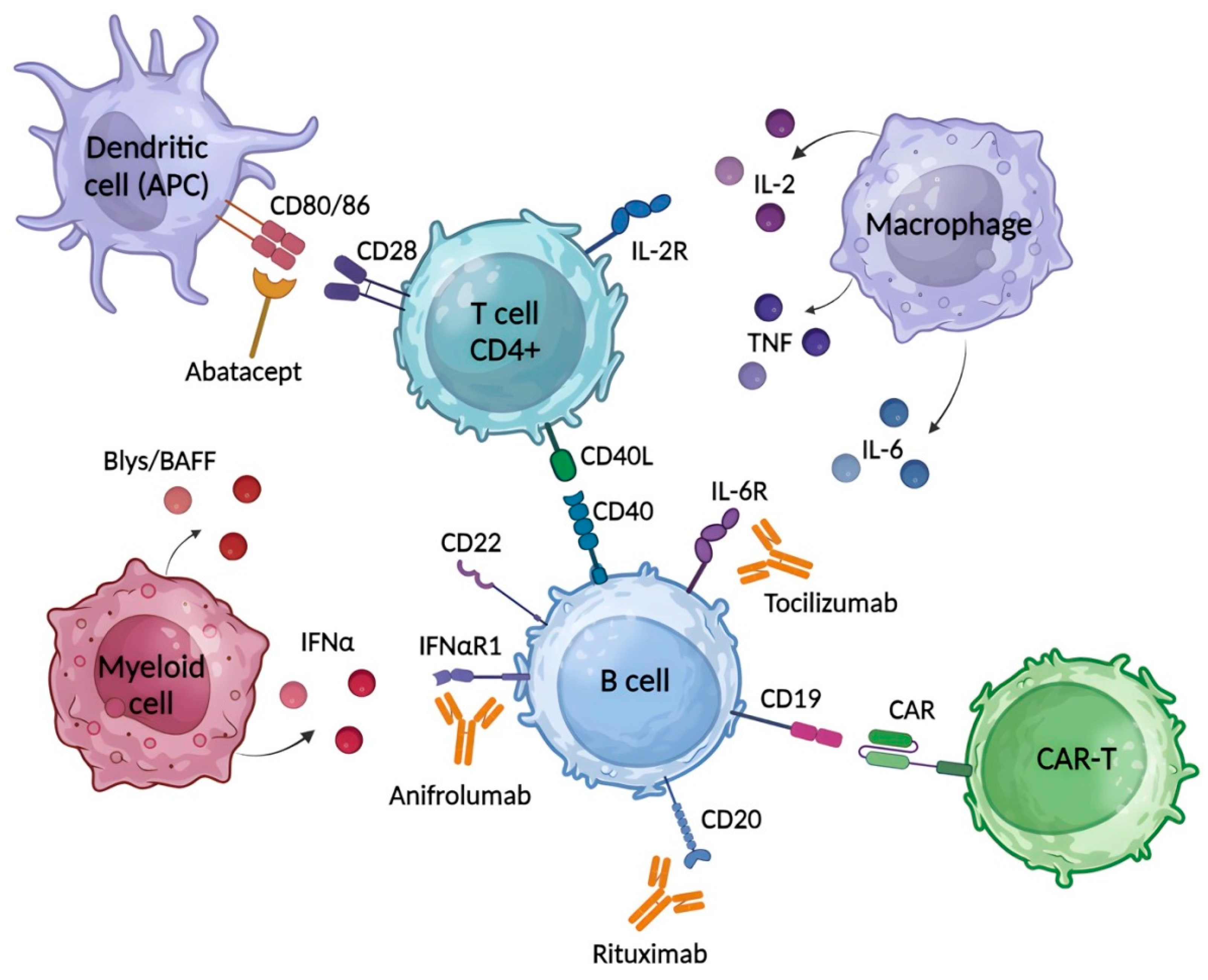{kind=link}
Abstract
:1. Introduction
2. Systemic Lupus Erythematosus (SLE)
3. Systemic Sclerosis (SSc)
4. Rheumatoid Arthritis (RA)
5. Common Therapeutics for SLE, SSc and RA
6. Conclusions and Perspective
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
List of Abbreviations
| ADCC | Antibody-dependent cell-mediated cytotoxicity |
| ACPA | Anticitrullinated protein antibody |
| APRIL | A proliferating inducing ligand |
| BAFF | B cell activating factor |
| BsAb | Bispecific antibody |
| CAR-T cell | Chimeric antigen receptor T cell |
| CAAR-T cell | Chimeric autoantibody receptor T cell |
| CCLE | Chronic cutaneous lupus erythematosus |
| CDC | Complement-dependent cytotoxicity |
| CLE | Cutaneous lupus erythematosus |
| CTLA-4 | Cytotoxic T lymphocyte-associated protein 4 |
| DLE | Discoid lupus erythematosus |
| FDA US | Food and Drug Administration |
| FITC | Fluorescein isothiocyanate |
| FLS | Fibroblast-like synoviocytes |
| GM-CSF | Granulocyte-macrophage colony-stimulating factor |
| JAK | Janus kinase family |
| mAb | Monoclonal antibody |
| SAIDs | Systemic autoimmune diseases |
| SLE | Systemic lupus erythematosus |
| TNF | Tumor necrosis factor |
| RA | Rheumatoid arthritis |
References
- Wang, L.; Wang, F.; Gershwin, M.E. Human Autoimmune Diseases: A Comprehensive Update. J. Intern. Med. 2015, 278, 369–395. [Google Scholar] [CrossRef]
- Dima, A.; Jurcut, C.; Arnaud, L. Hydroxychloroquine in Systemic and Autoimmune Diseases: Where Are We Now? Jt. Bone Spine 2021, 88, 105143. [Google Scholar] [CrossRef]
- Emamikia, S.; Gentline, C.; Chatzidionysiou, K.; Arnaud, L.; van Vollenhoven, R. Relationship between Glucocorticoid Dose and Adverse Events in Systemic Lupus Erythematosus: Data from a Randomized Clinical Trial. Scand. J. Rheumatol. 2018, 47, 131–140. [Google Scholar] [CrossRef]
- Dubey, A.K.; Handu, S.S.; Dubey, S.; Sharma, P.; Sharma, K.K. Belimumab: First Targeted Biological Treatment for Systemic Lupus Erythematosus. J. Pharmacol. Pharmacother. 2011, 2, 317–319. [Google Scholar] [CrossRef]
- Felten, R.; Dervovic, E.; Chasset, F.; Gottenberg, J.-E.; Sibilia, J.; Scher, F.; Arnaud, L. The 2018 Pipeline of Targeted Therapies under Clinical Development for Systemic Lupus Erythematosus: A Systematic Review of Trials. Autoimmun. Rev. 2018, 17, 781–790. [Google Scholar] [CrossRef]
- Hafeez, U.; Gan, H.K.; Scott, A.M. Monoclonal Antibodies as Immunomodulatory Therapy against Cancer and Autoimmune Diseases. Curr. Opin. Pharmacol. 2018, 41, 114–121. [Google Scholar] [CrossRef]
- Zhao, Q. Bispecific Antibodies for Autoimmune and Inflammatory Diseases: Clinical Progress to Date. BioDrugs 2020, 34, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Kalayci, F.N.C.; Ozen, S. Possible Role of Dysbiosis of the Gut Microbiome in SLE. Curr. Rheumatol. Rep. 2023, 25, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Pan, Q.; Guo, F.; Huang, Y.; Li, A.; Chen, S.; Chen, J.; Liu, H.; Pan, Q. Gut Microbiota Dysbiosis in Systemic Lupus Erythematosus: Novel Insights into Mechanisms and Promising Therapeutic Strategies. Front. Immunol. 2021, 12, 799788. [Google Scholar] [CrossRef] [PubMed]
- Mackensen, A.; Müller, F.; Mougiakakos, D.; Böltz, S.; Wilhelm, A.; Aigner, M.; Völkl, S.; Simon, D.; Kleyer, A.; Munoz, L.; et al. Anti-CD19 CAR T Cell Therapy for Refractory Systemic Lupus Erythematosus. Nat. Med. 2022, 28, 2124–2132. [Google Scholar] [CrossRef]
- Bergmann, C.; Müller, F.; Jörg, D.; Györfi, D.M.H.; Völkl, S.; Aigner, M.; Harrer, T.; Bayerl, N.; Atzinger, A.; Taubmann, J.; et al. AB0816 Treatment of a Patient with Severe Diffuse Systemic Sclerosis (SSC) Using CD19-Targeting CAR-T-Cells. In Scientific Abstracts; BMJ Publishing Group Ltd.: London, UK; European League Against Rheumatism: Zürich, Switzerland, 2023; p. 1621. [Google Scholar]
- Ganeeva, I.; Zmievskaya, E.; Valiullina, A.; Kudriaeva, A.; Miftakhova, R.; Rybalov, A.; Bulatov, E. Recent Advances in the Development of Bioreactors for Manufacturing of Adoptive Cell Immunotherapies. Bioengineering 2022, 9, 808. [Google Scholar] [CrossRef]
- Yaniv, G.; Twig, G.; Shor, D.B.-A.; Furer, A.; Sherer, Y.; Mozes, O.; Komisar, O.; Slonimsky, E.; Klang, E.; Lotan, E.; et al. A Volcanic Explosion of Autoantibodies in Systemic Lupus Erythematosus: A Diversity of 180 Different Antibodies Found in SLE Patients. Autoimmun. Rev. 2015, 14, 75–79. [Google Scholar] [CrossRef]
- Barber, M.R.W.; Drenkard, C.; Falasinnu, T.; Hoi, A.; Mak, A.; Kow, N.Y.; Svenungsson, E.; Peterson, J.; Clarke, A.E.; Ramsey-Goldman, R. Global Epidemiology of Systemic Lupus Erythematosus. Nat. Rev. Rheumatol. 2021, 17, 515–532. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Zhao, M.; Wu, H.; Lu, Q. A Comprehensive Review of Biological Agents for Lupus: Beyond Single Target. Front. Immunol. 2020, 11, 539797. [Google Scholar] [CrossRef]
- Mohan, C.; Putterman, C. Genetics and Pathogenesis of Systemic Lupus Erythematosus and Lupus Nephritis. Nat. Rev. Nephrol. 2015, 11, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Mok, M.Y.; Shoenfeld, Y. Recent Advances and Current State of Immunotherapy in Systemic Lupus Erythematosus. Expert Opin. Biol. Ther. 2016, 16, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Labrijn, A.F.; Janmaat, M.L.; Reichert, J.M.; Parren, P.W.H.I. Bispecific Antibodies: A Mechanistic Review of the Pipeline. Nat. Rev. Drug Discov. 2019, 18, 585–608. [Google Scholar] [CrossRef]
- Brinkmann, U.; Kontermann, R.E. The Making of Bispecific Antibodies. MAbs 2017, 9, 182–212. [Google Scholar] [CrossRef]
- Spiess, C.; Zhai, Q.; Carter, P.J. Alternative Molecular Formats and Therapeutic Applications for Bispecific Antibodies. Mol. Immunol. 2015, 67, 95–106. [Google Scholar] [CrossRef]
- Robinson, R. Distinct B Cell Receptor Functions Are Determined by Phosphorylation. PLoS Biol. 2006, 4, e231. [Google Scholar] [CrossRef]
- Tanaka, Y.; Kubo, S.; Iwata, S.; Yoshikawa, M.; Nakayamada, S. B Cell Phenotypes, Signaling and Their Roles in Secretion of Antibodies in Systemic Lupus Erythematosus. Clin. Immunol. 2018, 186, 21–25. [Google Scholar] [CrossRef]
- Bossen, C.; Schneider, P. BAFF, APRIL and Their Receptors: Structure, Function and Signaling. Semin. Immunol. 2006, 18, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Zollars, E.; Bienkowska, J.; Czerkowicz, J.; Allaire, N.; Ranger, A.M.; Magder, L.; Petri, M. BAFF (B Cell Activating Factor) Transcript Level in Peripheral Blood of Patients with SLE Is Associated with Same-Day Disease Activity as Well as Global Activity over the next Year. Lupus Sci. Med. 2015, 2, e000063. [Google Scholar] [CrossRef] [PubMed]
- Mackay, F.; Ambrose, C. The TNF Family Members BAFF and APRIL: The Growing Complexity. Cytokine Growth Factor Rev. 2003, 14, 311–324. [Google Scholar] [CrossRef] [PubMed]
- Vincent, F.B.; Morand, E.F.; Schneider, P.; Mackay, F. The BAFF/APRIL System in SLE Pathogenesis. Nat. Rev. Rheumatol. 2014, 10, 365–373. [Google Scholar] [CrossRef]
- Baker, K.P.; Edwards, B.M.; Main, S.H.; Choi, G.H.; Wager, R.E.; Halpern, W.G.; Lappin, P.B.; Riccobene, T.; Abramian, D.; Sekut, L.; et al. Generation and Characterization of LymphoStat-B, a Human Monoclonal Antibody That Antagonizes the Bioactivities of B Lymphocyte Stimulator. Arthritis Rheum. 2003, 48, 3253–3265. [Google Scholar] [CrossRef]
- Furie, R.; Petri, M.; Zamani, O.; Cervera, R.; Wallace, D.J.; Tegzová, D.; Sanchez-Guerrero, J.; Schwarting, A.; Merrill, J.T.; Chatham, W.W.; et al. A Phase III, Randomized, Placebo-Controlled Study of Belimumab, a Monoclonal Antibody That Inhibits B Lymphocyte Stimulator, in Patients with Systemic Lupus Erythematosus. Arthritis Rheum. 2011, 63, 3918–3930. [Google Scholar] [CrossRef]
- Navarra, S.V.; Guzmán, R.M.; Gallacher, A.E.; Hall, S.; Levy, R.A.; Jimenez, R.E.; Li, E.K.-M.; Thomas, M.; Kim, H.-Y.; León, M.G.; et al. Efficacy and Safety of Belimumab in Patients with Active Systemic Lupus Erythematosus: A Randomised, Placebo-Controlled, Phase 3 Trial. Lancet 2011, 377, 721–731. [Google Scholar] [CrossRef]
- Bezalel, S.; Guri, K.M.; Elbirt, D.; Asher, I.; Sthoeger, Z.M. Type I Interferon Signature in Systemic Lupus Erythematosus. Isr. Med. Assoc. J. 2014, 16, 246–249. [Google Scholar]
- Accapezzato, D.; Caccavale, R.; Paroli, M.P.; Gioia, C.; Nguyen, B.L.; Spadea, L.; Paroli, M. Advances in the Pathogenesis and Treatment of Systemic Lupus Erythematosus. Int. J. Mol. Sci. 2023, 24, 6578. [Google Scholar] [CrossRef]
- O’Shea, J.J.; Plenge, R. JAK and STAT Signaling Molecules in Immunoregulation and Immune-Mediated Disease. Immunity 2012, 36, 542–550. [Google Scholar] [CrossRef] [PubMed]
- de Veer, M.J.; Holko, M.; Frevel, M.; Walker, E.; Der, S.; Paranjape, J.M.; Silverman, R.H.; Williams, B.R. Functional Classification of Interferon-Stimulated Genes Identified Using Microarrays. J. Leukoc. Biol. 2001, 69, 912–920. [Google Scholar] [CrossRef] [PubMed]
- Christensen, S.R.; Shupe, J.; Nickerson, K.; Kashgarian, M.; Flavell, R.A.; Shlomchik, M.J. Toll-like Receptor 7 and TLR9 Dictate Autoantibody Specificity and Have Opposing Inflammatory and Regulatory Roles in a Murine Model of Lupus. Immunity 2006, 25, 417–428. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Tago, F.; Yamakawa, N.; Aoki, M.; Yagi, T.; Akira, S. A New Therapeutic Target for Systemic Lupus Erythematosus: The Current Landscape for Drug Development of a Toll-like Receptor 7/8 Antagonist through Academia-Industry-Government Collaboration. Immunol. Med. 2023, 1–6. [Google Scholar] [CrossRef]
- Dörner, T.; Lipsky, P.E. Beyond Pan-B-Cell-Directed Therapy—New Avenues and Insights into the Pathogenesis of SLE. Nat. Rev. Rheumatol. 2016, 12, 645–657. [Google Scholar] [CrossRef]
- Macauley, M.S.; Pfrengle, F.; Rademacher, C.; Nycholat, C.M.; Gale, A.J.; von Drygalski, A.; Paulson, J.C. Antigenic Liposomes Displaying CD22 Ligands Induce Antigen-Specific B Cell Apoptosis. J. Clin. Investig. 2013, 123, 3074–3083. [Google Scholar] [CrossRef]
- Ereño-Orbea, J.; Sicard, T.; Cui, H.; Mazhab-Jafari, M.T.; Benlekbir, S.; Guarné, A.; Rubinstein, J.L.; Julien, J.-P. Molecular Basis of Human CD22 Function and Therapeutic Targeting. Nat. Commun. 2017, 8, 764. [Google Scholar] [CrossRef]
- Du, F.H.; Mills, E.A.; Mao-Draayer, Y. Next-Generation Anti-CD20 Monoclonal Antibodies in Autoimmune Disease Treatment. Autoimmun. Highlights 2017, 8, 12. [Google Scholar] [CrossRef]
- Daridon, C.; Blassfeld, D.; Reiter, K.; Mei, H.E.; Giesecke, C.; Goldenberg, D.M.; Hansen, A.; Hostmann, A.; Frölich, D.; Dörner, T. Epratuzumab Targeting of CD22 Affects Adhesion Molecule Expression and Migration of B-Cells in Systemic Lupus Erythematosus. Arthritis Res. Ther. 2010, 12, R204. [Google Scholar] [CrossRef]
- Shipa, M.; Embleton-Thirsk, A.; Parvaz, M.; Santos, L.R.; Muller, P.; Chowdhury, K.; Isenberg, D.A.; Doré, C.J.; Gordon, C.; Ehrenstein, M.R. Effectiveness of Belimumab After Rituximab in Systemic Lupus Erythematosus. Ann. Intern. Med. 2021, 174, 1647–1657. [Google Scholar] [CrossRef]
- Li, X.; Ding, Y.; Zi, M.; Sun, L.; Zhang, W.; Chen, S.; Xu, Y. CD19, from Bench to Bedside. Immunol. Lett. 2017, 183, 86–95. [Google Scholar] [CrossRef]
- Espéli, M.; Smith, K.G.C.; Clatworthy, M.R. FcγRIIB and Autoimmunity. Immunol. Rev. 2016, 269, 194–211. [Google Scholar] [CrossRef]
- Horton, H.M.; Chu, S.Y.; Ortiz, E.C.; Pong, E.; Cemerski, S.; Leung, I.W.L.; Jacob, N.; Zalevsky, J.; Desjarlais, J.R.; Stohl, W.; et al. Antibody-Mediated Coengagement of FcγRIIb and B Cell Receptor Complex Suppresses Humoral Immunity in Systemic Lupus Erythematosus. J. Immunol. 2011, 186, 4223–4233. [Google Scholar] [CrossRef]
- Merrill, J.T.; Guthridge, J.; Smith, M.; June, J.; Koumpouras, F.; Machua, W.; Askanase, A.; Khosroshahi, A.; Sheikh, S.Z.; Rathi, G.; et al. A Double-Blind, Randomized, Placebo-Controlled, Phase 2 Trial of Obexelimab in Systemic Lupus Erythematosus with Exploration of Response Based on Gene Pathway Co-Expression Patterns. Arthritis Rheumatol. 2023, 75, 2185–2194. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.-Q.; Wang, J.-P.; Dai, Z.-W.; Mao, Y.-M.; Wu, J.; Guo, H.-S.; Xia, Y.-R.; Ye, D.-Q. Increased Circulating CXCL13 Levels in Systemic Lupus Erythematosus and Rheumatoid Arthritis: A Meta-Analysis. Clin. Rheumatol. 2020, 39, 281–290. [Google Scholar] [CrossRef]
- Klimatcheva, E.; Pandina, T.; Reilly, C.; Torno, S.; Bussler, H.; Scrivens, M.; Jonason, A.; Mallow, C.; Doherty, M.; Paris, M.; et al. CXCL13 Antibody for the Treatment of Autoimmune Disorders. BMC Immunol. 2015, 16, 6. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.-M.; Tsokos, G.C. T Cell Abnormalities in the Pathogenesis of Systemic Lupus Erythematosus: An Update. Curr. Rheumatol. Rep. 2021, 23, 12. [Google Scholar] [CrossRef] [PubMed]
- Shah, D.; Kiran, R.; Wanchu, A.; Bhatnagar, A. Oxidative Stress in Systemic Lupus Erythematosus: Relationship to Th1 Cytokine and Disease Activity. Immunol. Lett. 2010, 129, 7–12. [Google Scholar] [CrossRef]
- Akahoshi, M.; Nakashima, H.; Tanaka, Y.; Kohsaka, T.; Nagano, S.; Ohgami, E.; Arinobu, Y.; Yamaoka, K.; Niiro, H.; Shinozaki, M.; et al. Th1/Th2 Balance of Peripheral T Helper Cells in Systemic Lupus Erythematosus. Arthritis Rheum. 1999, 42, 1644–1648. [Google Scholar] [CrossRef]
- Sugimoto, K.; Morimoto, S.; Kaneko, H.; Nozawa, K.; Tokano, Y.; Takasaki, Y.; Hashimoto, H. Decreased IL-4 Producing CD4+ T Cells in Patients with Active Systemic Lupus Erythematosus-Relation to IL-12R Expression. AutoImmunity 2002, 35, 381–387. [Google Scholar] [CrossRef]
- Paroli, M.; Caccavale, R.; Fiorillo, M.T.; Spadea, L.; Gumina, S.; Candela, V.; Paroli, M.P. The Double Game Played by Th17 Cells in Infection: Host Defense and Immunopathology. Pathogens 2022, 11, 1547. [Google Scholar] [CrossRef] [PubMed]
- López, P.; Rodríguez-Carrio, J.; Caminal-Montero, L.; Mozo, L.; Suárez, A. A Pathogenic IFNα, BLyS and IL-17 Axis in Systemic Lupus Erythematosus Patients. Sci. Rep. 2016, 6, 20651. [Google Scholar] [CrossRef] [PubMed]
- Zickert, A.; Amoudruz, P.; Sundström, Y.; Rönnelid, J.; Malmström, V.; Gunnarsson, I. IL-17 and IL-23 in Lupus Nephritis—Association to Histopathology and Response to Treatment. BMC Immunol. 2015, 16, 7. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Luo, L.; Wu, Y.; Song, Z.; Ni, B.; Hao, F.; Luo, N. Elevated Apoptosis and Abnormal Apoptosis Signaling of Regulatory T Cells in Patients with Systemic Lupus Erythematosus. Lupus 2022, 31, 1441–1455. [Google Scholar] [CrossRef]
- Tsai, Y.-G.; Liao, P.-F.; Hsiao, K.-H.; Wu, H.-M.; Lin, C.-Y.; Yang, K.D. Pathogenesis and Novel Therapeutics of Regulatory T Cell Subsets and Interleukin-2 Therapy in Systemic Lupus Erythematosus. Front. Immunol. 2023, 14, 1230264. [Google Scholar] [CrossRef]
- Miao, M.; Xiao, X.; Tian, J.; Zhufeng, Y.; Feng, R.; Zhang, R.; Chen, J.; Zhang, X.; Huang, B.; Jin, Y.; et al. Therapeutic Potential of Targeting Tfr/Tfh Cell Balance by Low-Dose-IL-2 in Active SLE: A Post Hoc Analysis from a Double-Blind RCT Study. Arthritis Res. Ther. 2021, 23, 167. [Google Scholar] [CrossRef]
- He, J.; Zhang, R.; Shao, M.; Zhao, X.; Miao, M.; Chen, J.; Liu, J.; Zhang, X.; Zhang, X.; Jin, Y.; et al. Efficacy and Safety of Low-Dose IL-2 in the Treatment of Systemic Lupus Erythematosus: A Randomised, Double-Blind, Placebo-Controlled Trial. Ann. Rheum. Dis. 2020, 79, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Koenig, K.F.; Groeschl, I.; Pesickova, S.S.; Tesar, V.; Eisenberger, U.; Trendelenburg, M. Serum Cytokine Profile in Patients with Active Lupus Nephritis. Cytokine 2012, 60, 410–416. [Google Scholar] [CrossRef]
- Mok, M.Y.; Wu, H.J.; Lo, Y.; Lau, C.S. The Relation of Interleukin 17 (IL-17) and IL-23 to Th1/Th2 Cytokines and Disease Activity in Systemic Lupus Erythematosus. J. Rheumatol. 2010, 37, 2046–2052. [Google Scholar] [CrossRef]
- Mok, M.Y.; Huang, F.P.; Ip, W.K.; Lo, Y.; Wong, F.Y.; Chan, E.Y.T.; Lam, K.F.; Xu, D. Serum Levels of IL-33 and Soluble ST2 and Their Association with Disease Activity in Systemic Lupus Erythematosus. Rheumatology 2010, 49, 520–527. [Google Scholar] [CrossRef]
- Merrill, J.T.; Neuwelt, C.M.; Wallace, D.J.; Shanahan, J.C.; Latinis, K.M.; Oates, J.C.; Utset, T.O.; Gordon, C.; Isenberg, D.A.; Hsieh, H.-J.; et al. Efficacy and Safety of Rituximab in Moderately-to-Severely Active Systemic Lupus Erythematosus: The Randomized, Double-Blind, Phase Ii/Iii Systemic Lupus Erythematosus Evaluation of Rituximab Trial. Arthritis Rheum. 2010, 62, 222–233. [Google Scholar] [CrossRef] [PubMed]
- Kamburova, E.G.; Koenen, H.J.P.M.; Borgman, K.J.E.; ten Berge, I.J.; Joosten, I.; Hilbrands, L.B. A Single Dose of Rituximab Does Not Deplete B Cells in Secondary Lymphoid Organs but Alters Phenotype and Function. Am. J. Transplant. 2013, 13, 1503–1511. [Google Scholar] [CrossRef]
- Tedder, T.F.; Engel, P. CD20: A Regulator of Cell-Cycle Progression of B Lymphocytes. Immunol. Today 1994, 15, 450–454. [Google Scholar] [CrossRef]
- June, C.H.; Sadelain, M. Chimeric Antigen Receptor Therapy. N. Engl. J. Med. 2018, 379, 64–73. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Restifo, N.P. Adoptive Cell Transfer as Personalized Immunotherapy for Human Cancer. Science 2015, 348, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Valiullina, A.K.; Zmievskaya, E.A.; Ganeeva, I.A.; Zhuravleva, M.N.; Garanina, E.E.; Rizvanov, A.A.; Petukhov, A.V.; Bulatov, E.R. Evaluation of CAR-T Cells’ Cytotoxicity against Modified Solid Tumor Cell Lines. Biomedicines 2023, 11, 626. [Google Scholar] [CrossRef] [PubMed]
- Kansal, R.; Richardson, N.; Neeli, I.; Khawaja, S.; Chamberlain, D.; Ghani, M.; Ghani, Q.; Balazs, L.; Beranova-Giorgianni, S.; Giorgianni, F.; et al. Sustained B Cell Depletion by CD19-Targeted CAR T Cells Is a Highly Effective Treatment for Murine Lupus. Sci. Transl. Med. 2019, 11, eaav1648. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Xu, Q.; Pu, C.; Zhu, K.; Lu, C.; Jiang, Y.; Xiao, L.; Han, Y.; Lu, L. Therapeutic Efficacy of Anti-CD19 CAR-T Cells in a Mouse Model of Systemic Lupus Erythematosus. Cell. Mol. Immunol. 2021, 18, 1896–1903. [Google Scholar] [CrossRef]
- Mougiakakos, D.; Krönke, G.; Völkl, S.; Kretschmann, S.; Aigner, M.; Kharboutli, S.; Böltz, S.; Manger, B.; Mackensen, A.; Schett, G. CD19-Targeted CAR T Cells in Refractory Systemic Lupus Erythematosus. N. Engl. J. Med. 2021, 385, 567–569. [Google Scholar] [CrossRef]
- Boulougoura, A.; Gendelman, H.; Surmachevska, N.; Kyttaris, V.C. Journal Club: Anti-CD19 Chimeric Antigen Receptor T Cell Therapy for Refractory Systemic Lupus Erythematosus. ACR Open Rheumatol. 2023, 5, 624–628. [Google Scholar] [CrossRef]
- Zhang, W.; Feng, J.; Cinquina, A.; Wang, Q.; Xu, H.; Zhang, Q.; Sun, L.; Chen, Q.; Xu, L.; Pinz, K.; et al. Treatment of Systemic Lupus Erythematosus Using BCMA-CD19 Compound CAR. Stem Cell Rev. Rep. 2021, 17, 2120–2123. [Google Scholar] [CrossRef]
- Santamaria-Alza, Y.; Vasquez, G. Are Chimeric Antigen Receptor T Cells (CAR-T Cells) the Future in Immunotherapy for Autoimmune Diseases? Inflamm. Res. 2021, 70, 651–663. [Google Scholar] [CrossRef]
- Ellebrecht, C.T.; Bhoj, V.G.; Nace, A.; Choi, E.J.; Mao, X.; Cho, M.J.; Di Zenzo, G.; Lanzavecchia, A.; Seykora, J.T.; Cotsarelis, G.; et al. Reengineering Chimeric Antigen Receptor T Cells for Targeted Therapy of Autoimmune Disease. Science 2016, 353, 179–184. [Google Scholar] [CrossRef]
- Reincke, S.M.; von Wardenburg, N.; Homeyer, M.A.; Kornau, H.-C.; Spagni, G.; Li, L.Y.; Kreye, J.; Sánchez-Sendín, E.; Blumenau, S.; Stappert, D.; et al. Chimeric Autoantibody Receptor T Cells Deplete NMDA Receptor-Specific B Cells. Cell 2023, 186, 5084–5097.e18. [Google Scholar] [CrossRef]
- Li, Y.-J.; Chen, Z. Cell-Based Therapies for Rheumatoid Arthritis: Opportunities and Challenges. Ther. Adv. Musculoskelet. Dis. 2022, 14, 1759720X2211002. [Google Scholar] [CrossRef]
- Tenspolde, M.; Zimmermann, K.; Weber, L.C.; Hapke, M.; Lieber, M.; Dywicki, J.; Frenzel, A.; Hust, M.; Galla, M.; Buitrago-Molina, L.E.; et al. Regulatory T Cells Engineered with a Novel Insulin-Specific Chimeric Antigen Receptor as a Candidate Immunotherapy for Type 1 Diabetes. J. Autoimmun. 2019, 103, 102289. [Google Scholar] [CrossRef]
- Kaminskiy, Y.; Kuznetsova, V.; Kudriaeva, A.; Zmievskaya, E.; Bulatov, E. Neglected, yet Significant Role of FOXP1 in T-Cell Quiescence, Differentiation and Exhaustion. Front. Immunol. 2022, 13, 971045. [Google Scholar] [CrossRef] [PubMed]
- Zmievskaya, E.; Valiullina, A.; Ganeeva, I.; Petukhov, A.; Rizvanov, A.; Bulatov, E. Application of CAR-T Cell Therapy beyond Oncology: Autoimmune Diseases and Viral Infections. Biomedicines 2021, 9, 59. [Google Scholar] [CrossRef] [PubMed]
- Boroughs, A.C.; Larson, R.C.; Choi, B.D.; Bouffard, A.A.; Riley, L.S.; Schiferle, E.; Kulkarni, A.S.; Cetrulo, C.L.; Ting, D.; Blazar, B.R.; et al. Chimeric Antigen Receptor Costimulation Domains Modulate Human Regulatory T Cell Function. JCI Insight 2019, 4, e126194. [Google Scholar] [CrossRef] [PubMed]
- Műzes, G.; Sipos, F. CAR-Based Therapy for Autoimmune Diseases: A Novel Powerful Option. Cells 2023, 12, 1534. [Google Scholar] [CrossRef] [PubMed]
- Pattanaik, D.; Brown, M.; Postlethwaite, B.C.; Postlethwaite, A.E. Pathogenesis of Systemic Sclerosis. Front. Immunol. 2015, 6, 272. [Google Scholar] [CrossRef]
- Sato, S.; Fujimoto, M.; Hasegawa, M.; Takehara, K. Altered Blood B Lymphocyte Homeostasis in Systemic Sclerosis: Expanded Naive B Cells and Diminished but Activated Memory B Cells. Arthritis Rheum. 2004, 50, 1918–1927. [Google Scholar] [CrossRef]
- Gordon, J.K.; Martyanov, V.; Franks, J.M.; Bernstein, E.J.; Szymonifka, J.; Magro, C.; Wildman, H.F.; Wood, T.A.; Whitfield, M.L.; Spiera, R.F. Belimumab for the Treatment of Early Diffuse Systemic Sclerosis. Arthritis Rheumatol. 2018, 70, 308–316. [Google Scholar] [CrossRef]
- Giacomelli, R.; Cipriani, P.; Lattanzio, R.; DI Franco, M.; Locanto, M.; Parzanese, I.; Passacantando, A.; Ciocci, A.; Tonietti, G. Circulating Levels of Soluble CD30 Are Increased in Patients with Systemic Sclerosis (SSc) and Correlate with Serological and Clinical Features of the Disease. Clin. Exp. Immunol. 2003, 108, 42–46. [Google Scholar] [CrossRef]
- Farina, N.; Campochiaro, C.; Lescoat, A.; Benanti, G.; De Luca, G.; Khanna, D.; Dagna, L.; Matucci-Cerinic, M. Drug Development and Novel Therapeutics to Ensure a Personalized Approach in the Treatment of Systemic Sclerosis. Expert Rev. Clin. Immunol. 2023, 19, 1131–1142. [Google Scholar] [CrossRef] [PubMed]
- Mulcaire-Jones, E.; Low, A.H.L.; Domsic, R.; Whitfield, M.L.; Khanna, D. Advances in Biological and Targeted Therapies for Systemic Sclerosis. Expert Opin. Biol. Ther. 2023, 23, 325–339. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, S.; Cant, R.; Ciechomska, M.; van Laar, J.M. Interleukin-6: A New Therapeutic Target in Systemic Sclerosis? Clin. Transl. Immunol. 2013, 2, e4. [Google Scholar] [CrossRef]
- Derynck, R.; Budi, E.H. Specificity, Versatility, and Control of TGF-β Family Signaling. Sci. Signal. 2019, 12, eaav5183. [Google Scholar] [CrossRef]
- Wei, L.; Abraham, D.; Ong, V. The Yin and Yang of IL-17 in Systemic Sclerosis. Front. Immunol. 2022, 13, 885609. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Mu, R.; Wei, X. The Roles of IL-1 Family Cytokines in the Pathogenesis of Systemic Sclerosis. Front. Immunol. 2019, 10, 2025. [Google Scholar] [CrossRef]
- Kuzumi, A.; Yoshizaki, A.; Matsuda, K.M.; Kotani, H.; Norimatsu, Y.; Fukayama, M.; Ebata, S.; Fukasawa, T.; Yoshizaki-Ogawa, A.; Asano, Y.; et al. Interleukin-31 Promotes Fibrosis and T Helper 2 Polarization in Systemic Sclerosis. Nat. Commun. 2021, 12, 5947. [Google Scholar] [CrossRef] [PubMed]
- Kakkar, V.; Assassi, S.; Allanore, Y.; Kuwana, M.; Denton, C.P.; Khanna, D.; Del Galdo, F. Type 1 Interferon Activation in Systemic Sclerosis: A Biomarker, a Target or the Culprit. Curr. Opin. Rheumatol. 2022, 34, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Komura, K.; Yoshizaki, A.; Kodera, M.; Iwata, Y.; Ogawa, F.; Shumizu, K.; Wayaki, T.; Yukami, T.; Murata, M.; Hasegawa, M.; et al. Increased Serum Soluble OX40 in Patients with Systemic Sclerosis. J. Rheumatol. 2008, 35, 2359–2362. [Google Scholar] [CrossRef] [PubMed]
- Senoo, S.; Taniguchi, A.; Itano, J.; Oda, N.; Morichika, D.; Fujii, U.; Guo, L.; Sunami, R.; Kanehiro, A.; Tokioka, F.; et al. Essential Role of IL-23 in the Development of Acute Exacerbation of Pulmonary Fibrosis. Am. J. Physiol.-Lung. Cell. Mol. Physiol. 2021, 321, L925–L940. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, C.; Müller, F.; Distler, J.H.W.; Györfi, A.-H.; Völkl, S.; Aigner, M.; Kretschmann, S.; Reimann, H.; Harrer, T.; Bayerl, N.; et al. Treatment of a Patient with Severe Systemic Sclerosis (SSc) Using CD19-Targeted CAR T Cells. Ann. Rheum. Dis. 2023, 82, 1117–1120. [Google Scholar] [CrossRef]
- Huang, J.; Fu, X.; Chen, X.; Li, Z.; Huang, Y.; Liang, C. Promising Therapeutic Targets for Treatment of Rheumatoid Arthritis. Front. Immunol. 2021, 12, 686155. [Google Scholar] [CrossRef]
- Lin, Y.-J.; Anzaghe, M.; Schülke, S. Update on the Pathomechanism, Diagnosis, and Treatment Options for Rheumatoid Arthritis. Cells 2020, 9, 880. [Google Scholar] [CrossRef]
- Nygaard, G.; Firestein, G.S. Restoring Synovial Homeostasis in Rheumatoid Arthritis by Targeting Fibroblast-like Synoviocytes. Nat. Rev. Rheumatol. 2020, 16, 316–333. [Google Scholar] [CrossRef]
- Kishimoto, T.; Kang, S. IL-6 Revisited: From Rheumatoid Arthritis to CAR T Cell Therapy and COVID-19. Annu. Rev. Immunol. 2022, 40, 323–348. [Google Scholar] [CrossRef]
- Barnas, J.L.; Looney, R.J.; Anolik, J.H. B Cell Targeted Therapies in Autoimmune Disease. Curr. Opin. Immunol. 2019, 61, 92–99. [Google Scholar] [CrossRef]
- Edwards, J.C.W.; Szczepański, L.; Szechiński, J.; Filipowicz-Sosnowska, A.; Emery, P.; Close, D.R.; Stevens, R.M.; Shaw, T. Efficacy of B-Cell–Targeted Therapy with Rituximab in Patients with Rheumatoid Arthritis. N. Engl. J. Med. 2004, 350, 2572–2581. [Google Scholar] [CrossRef]
- Kaplanski, G. IL-6: A Regulator of the Transition from Neutrophil to Monocyte Recruitment during Inflammation. Trends Immunol. 2003, 24, 25–29. [Google Scholar] [CrossRef]
- Hashizume, M.; Hayakawa, N.; Mihara, M. IL-6 Trans-Signalling Directly Induces RANKL on Fibroblast-like Synovial Cells and Is Involved in RANKL Induction by TNF- and IL-17. Rheumatology 2008, 47, 1635–1640. [Google Scholar] [CrossRef]
- Genovese, M.C.; Fleischmann, R.; Kivitz, A.J.; Rell-Bakalarska, M.; Martincova, R.; Fiore, S.; Rohane, P.; van Hoogstraten, H.; Garg, A.; Fan, C.; et al. Sarilumab Plus Methotrexate in Patients With Active Rheumatoid Arthritis and Inadequate Response to Methotrexate: Results of a Phase III Study. Arthritis Rheumatol. 2015, 67, 1424–1437. [Google Scholar] [CrossRef] [PubMed]
- Schaible, T.F. Long Term Safety of Infliximab. Can. J. Gastroenterol. 2000, 14, 29C–32C. [Google Scholar] [CrossRef]
- Rooney, M.; Symons, J.A.; Duff, G.W. Interleukin 1 Beta in Synovial Fluid Is Related to Local Disease Activity in Rheumatoid Arthritis. Rheumatol. Int. 1990, 10, 217–219. [Google Scholar] [CrossRef]
- Mertens, M.; Singh, J.A. Anakinra for Rheumatoid Arthritis: A Systematic Review. J. Rheumatol. 2009, 36, 1118–1125. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Wang, Y.; Yuan, Y.; Sun, J.; Liu, L.; Huang, D.; Hu, J.; Wang, M.; Li, S.; Song, W.; et al. In Vitro Elimination of Autoreactive B Cells from Rheumatoid Arthritis Patients by Universal Chimeric Antigen Receptor T Cells. Ann. Rheum. Dis. 2021, 80, 176–184. [Google Scholar] [CrossRef]
- Minutolo, N.G.; Hollander, E.E.; Powell, D.J. The Emergence of Universal Immune Receptor T Cell Therapy for Cancer. Front. Oncol. 2019, 9, 176. [Google Scholar] [CrossRef] [PubMed]
- Sakkas, L.I.; Bogdanos, D.P.; Katsiari, C.; Platsoucas, C.D. Anti-Citrullinated Peptides as Autoantigens in Rheumatoid Arthritis—Relevance to Treatment. Autoimmun. Rev. 2014, 13, 1114–1120. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Ma, J.S.Y.; Yun, H.; Cao, Y.; Kim, J.Y.; Chi, V.; Wang, D.; Woods, A.; Sherwood, L.; Caballero, D.; et al. Redirection of Genetically Engineered CAR-T Cells Using Bifunctional Small Molecules. J. Am. Chem. Soc. 2015, 137, 2832–2835. [Google Scholar] [CrossRef]
- Kaegi, C.; Wuest, B.; Schreiner, J.; Steiner, U.C.; Vultaggio, A.; Matucci, A.; Crowley, C.; Boyman, O. Systematic Review of Safety and Efficacy of Rituximab in Treating Immune-Mediated Disorders. Front. Immunol. 2019, 10, 1990. [Google Scholar] [CrossRef]
- Maher, T.M.; Tudor, V.A.; Saunders, P.; Gibbons, M.A.; Fletcher, S.V.; Denton, C.P.; Hoyles, R.K.; Parfrey, H.; Renzoni, E.A.; Kokosi, M.; et al. Rituximab versus Intravenous Cyclophosphamide in Patients with Connective Tissue Disease-Associated Interstitial Lung Disease in the UK (RECITAL): A Double-Blind, Double-Dummy, Randomised, Controlled, Phase 2b Trial. Lancet Respir. Med. 2023, 11, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Ebata, S.; Yoshizaki, A.; Oba, K.; Kashiwabara, K.; Ueda, K.; Uemura, Y.; Watadani, T.; Fukasawa, T.; Miura, S.; Yoshizaki-Ogawa, A.; et al. Safety and Efficacy of Rituximab in Systemic Sclerosis (DESIRES): A Double-Blind, Investigator-Initiated, Randomised, Placebo-Controlled Trial. Lancet Rheumatol. 2021, 3, e489–e497. [Google Scholar] [CrossRef] [PubMed]
- Morgan, K.; Woollard, C.; Beinart, D.; Host, L.V.; Roddy, J. Rituximab Treatment for Systemic Sclerosis-associated Interstitial Lung Disease: A Case Series of 13 Patients. Intern. Med. J. 2023, 53, 1147–1153. [Google Scholar] [CrossRef] [PubMed]
- Yoshifuji, H.; Yomono, K.; Yamano, Y.; Kondoh, Y.; Yasuoka, H. Role of Rituximab in the Treatment of Systemic Sclerosis: A Literature Review. Mod. Rheumatol. 2023, 33, 1068–1077. [Google Scholar] [CrossRef] [PubMed]
- Rowshanravan, B.; Halliday, N.; Sansom, D.M. CTLA-4: A Moving Target in Immunotherapy. Blood 2018, 131, 58–67. [Google Scholar] [CrossRef]
- Pombo-Suarez, M.; Gomez-Reino, J.J. Abatacept for the Treatment of Rheumatoid Arthritis. Expert Rev. Clin. Immunol. 2019, 15, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Pimentel-Quiroz, V.R.; Ugarte-Gil, M.F.; Alarcón, G.S. Abatacept for the Treatment of Systemic Lupus Erythematosus. Expert Opin. Investig. Drugs 2016, 25, 493–499. [Google Scholar] [CrossRef]
- Chung, L.; Spino, C.; McLain, R.; Johnson, S.R.; Denton, C.P.; Molitor, J.A.; Steen, V.D.; Lafyatis, R.; Simms, R.W.; Kafaja, S.; et al. Safety and Efficacy of Abatacept in Early Diffuse Cutaneous Systemic Sclerosis (ASSET): Open-Label Extension of a Phase 2, Double-Blind Randomised Trial. Lancet Rheumatol. 2020, 2, e743–e753. [Google Scholar] [CrossRef]
- Khanna, D.; Spino, C.; Johnson, S.; Chung, L.; Whitfield, M.L.; Denton, C.P.; Berrocal, V.; Franks, J.; Mehta, B.; Molitor, J.; et al. Abatacept in Early Diffuse Cutaneous Systemic Sclerosis: Results of a Phase II Investigator-Initiated, Multicenter, Double-Blind, Randomized, Placebo-Controlled Trial. Arthritis Rheumatol. 2020, 72, 125–136. [Google Scholar] [CrossRef]
- Lin, C.M.A.; Isaacs, J.D.; Cooles, F.A.H. Role of IFN-α in Rheumatoid Arthritis. Curr. Rheumatol. Rep. 2024, 26, 37–52. [Google Scholar] [CrossRef]
- Casey, K.A.; Guo, X.; Smith, M.A.; Wang, S.; Sinibaldi, D.; Sanjuan, M.A.; Wang, L.; Illei, G.G.; White, W.I. Type I Interferon Receptor Blockade with Anifrolumab Corrects Innate and Adaptive Immune Perturbations of SLE. Lupus Sci. Med. 2018, 5, e000286. [Google Scholar] [CrossRef]
- Liu, Z.; Cheng, R.; Liu, Y. Evaluation of Anifrolumab Safety in Systemic Lupus Erythematosus: A Meta-Analysis and Systematic Review. Front. Immunol. 2022, 13, 996662. [Google Scholar] [CrossRef]
- Tovey, M.G.; Lallemand, C. Immunogenicity and Other Problems Associated with the Use of Biopharmaceuticals. Ther. Adv. Drug Saf. 2011, 2, 113–128. [Google Scholar] [CrossRef] [PubMed]
- Maini, R.N.; Taylor, P.C.; Szechinski, J.; Pavelka, K.; Bröll, J.; Balint, G.; Emery, P.; Raemen, F.; Petersen, J.; Smolen, J.; et al. Double-blind Randomized Controlled Clinical Trial of the Interleukin-6 Receptor Antagonist, Tocilizumab, in European Patients with Rheumatoid Arthritis Who Had an Incomplete Response to Methotrexate. Arthritis Rheum. 2006, 54, 2817–2829. [Google Scholar] [CrossRef] [PubMed]
- Gabay, C.; Emery, P.; van Vollenhoven, R.; Dikranian, A.; Alten, R.; Pavelka, K.; Klearman, M.; Musselman, D.; Agarwal, S.; Green, J.; et al. Tocilizumab Monotherapy versus Adalimumab Monotherapy for Treatment of Rheumatoid Arthritis (ADACTA): A Randomised, Double-Blind, Controlled Phase 4 Trial. Lancet 2013, 381, 1541–1550. [Google Scholar] [CrossRef] [PubMed]
- Khanna, D.; Lin, C.J.F.; Furst, D.E.; Wagner, B.; Zucchetto, M.; Raghu, G.; Martinez, F.J.; Goldin, J.; Siegel, J.; Denton, C.P. Long-Term Safety and Efficacy of Tocilizumab in Early Systemic Sclerosis–Interstitial Lung Disease: Open-Label Extension of a Phase 3 Randomized Controlled Trial. Am. J. Respir. Crit. Care Med. 2022, 205, 674–684. [Google Scholar] [CrossRef] [PubMed]
- Roofeh, D.; Lin, C.J.F.; Goldin, J.; Kim, G.H.; Furst, D.E.; Denton, C.P.; Huang, S.; Khanna, D. Tocilizumab Prevents Progression of Early Systemic Sclerosis–Associated Interstitial Lung Disease. Arthritis Rheumatol. 2021, 73, 1301–1310. [Google Scholar] [CrossRef]
- Chaoyi, M.; Shrestha, B.; Hui, L.; Qiujin, D.; Ping, F. Tocilizumab Therapy for Persistent High-Grade Fever in Systemic Lupus Erythematosus: Two Cases and a Literature Review. J. Int. Med. Res. 2022, 50, 030006052210885. [Google Scholar] [CrossRef]
- Yadav, S.; Sharma, V.; Balakrishnan, C. Tocilizumab Therapy for Treatment-Resistant Systemic Lupus Erythematosus with Elevated IL-6 and CRP Levels: A Case Report. SN Compr. Clin. Med. 2023, 5, 199. [Google Scholar] [CrossRef]
- Kuriakose, A.; Chirmule, N.; Nair, P. Immunogenicity of Biotherapeutics: Causes and Association with Posttranslational Modifications. J. Immunol. Res. 2016, 2016, 1–18. [Google Scholar] [CrossRef]
- Mullard, A. CAR T Cell Therapies Raise Hopes—And Questions—For Lupus and Autoimmune Disease. Nat. Rev. Drug Discov. 2023, 22, 859–861. [Google Scholar] [CrossRef] [PubMed]
- Chasov, V.; Zaripov, M.; Mirgayazova, R.; Khadiullina, R.; Zmievskaya, E.; Ganeeva, I.; Valiullina, A.; Rizvanov, A.; Bulatov, E. Promising New Tools for Targeting P53 Mutant Cancers: Humoral and Cell-Based Immunotherapies. Front. Immunol. 2021, 12, 707734. [Google Scholar] [CrossRef] [PubMed]
- Chames, P.; Van Regenmortel, M.; Weiss, E.; Baty, D. Therapeutic Antibodies: Successes, Limitations and Hopes for the Future. Br. J. Pharmacol. 2009, 157, 220–233. [Google Scholar] [CrossRef] [PubMed]
- Almagro, J.C.; Daniels-Wells, T.R.; Perez-Tapia, S.M.; Penichet, M.L. Progress and Challenges in the Design and Clinical Development of Antibodies for Cancer Therapy. Front. Immunol. 2018, 8, 1751. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S. CAR-T Efficacy: Is Conditioning the Key? Blood 2019, 133, 1799–1800. [Google Scholar] [CrossRef]
- Sun, Y.; Yuan, Y.; Zhang, B.; Zhang, X. CARs: A New Approach for the Treatment of Autoimmune Diseases. Sci. China Life Sci. 2023, 66, 711–728. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chasov, V.; Zmievskaya, E.; Ganeeva, I.; Gilyazova, E.; Davletshin, D.; Khaliulin, M.; Kabwe, E.; Davidyuk, Y.N.; Valiullina, A.; Rizvanov, A.; et al. Immunotherapy Strategy for Systemic Autoimmune Diseases: Betting on CAR-T Cells and Antibodies. Antibodies 2024, 13, 10. https://doi.org/10.3390/antib13010010
Chasov V, Zmievskaya E, Ganeeva I, Gilyazova E, Davletshin D, Khaliulin M, Kabwe E, Davidyuk YN, Valiullina A, Rizvanov A, et al. Immunotherapy Strategy for Systemic Autoimmune Diseases: Betting on CAR-T Cells and Antibodies. Antibodies. 2024; 13(1):10. https://doi.org/10.3390/antib13010010
Chicago/Turabian StyleChasov, Vitaly, Ekaterina Zmievskaya, Irina Ganeeva, Elvina Gilyazova, Damir Davletshin, Marat Khaliulin, Emmanuel Kabwe, Yuriy N. Davidyuk, Aygul Valiullina, Albert Rizvanov, and et al. 2024. "Immunotherapy Strategy for Systemic Autoimmune Diseases: Betting on CAR-T Cells and Antibodies" Antibodies 13, no. 1: 10. https://doi.org/10.3390/antib13010010