
Characterization of N-Terminal Asparagine Deamidation and Clipping of a Monoclonal Antibody

Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Degraded Samples Preparation
2.3. IEC Chromatography and Fraction Collection
2.4. Imaged Capillary Isoelectric Focusing
2.5. Intact Mass Analysis
2.6. Tryptic Peptide Mapping
2.7. Lys-C Peptide Mapping
2.8. Potency Assay
3. Results
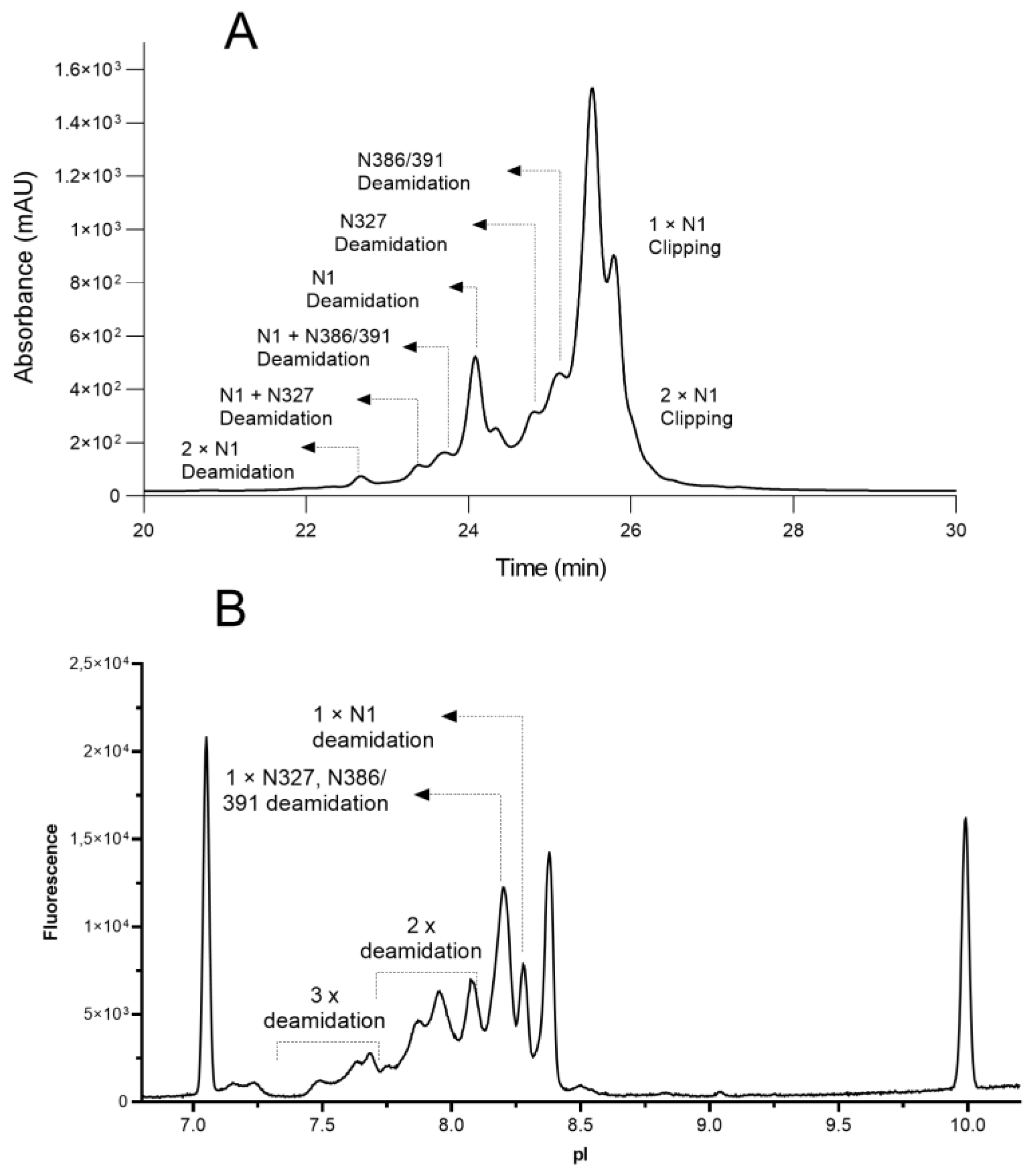
3.1. Charge Variants Separations
3.2. Charge Variants Identification
3.3. Protein Stability and Potency Impact
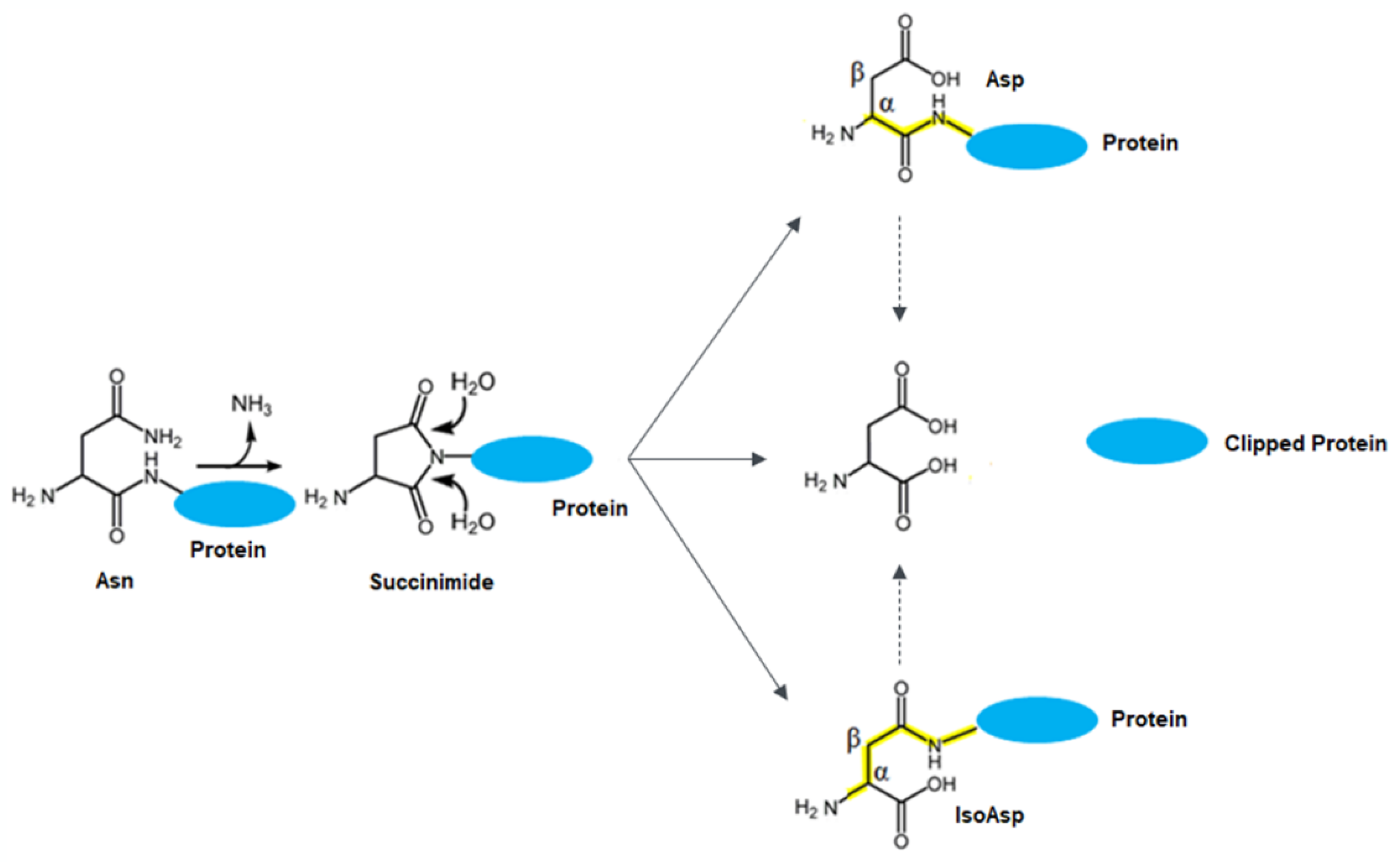
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cacia, J.; Keck, R.; Presta, L.G.; Frenz, J. Isomerization of an aspartic acid residue in the complementarity-determining regions of a recombinant antibody to human IgE: Identification and effect on binding affinity. Biochemistry 1996, 35, 1897–1903. [Google Scholar] [CrossRef] [PubMed]
- Ouellette, D.; Chumsae, C.; Clabbers, A.; Radziejewski, C.; Correia, I. Comparison of the in vitro and in vivo stability of a succinimide intermediate observed on a therapeutic IgG1 molecule. mAbs 2013, 5, 432–444. [Google Scholar] [CrossRef] [PubMed]
- Bults, P.; Bischoff, R.; Bakker, H.; Gietema, J.A.; van de Merbel, N.C. LC-MS/MS-Based Monitoring of In Vivo Protein Biotransformation: Quantitative Determination of Trastuzumab and Its Deamidation Products in Human Plasma. Anal. Chem. 2016, 88, 1871–1877. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Machiesky, L.A.; De Mel, N.; Du, Q.; Xu, W.; Washabaugh, M.; Jiang, X.-R.; Wang, J. Characterization of IgG1 Fc Deamidation at Asparagine 325 and Its Impact on Antibody-dependent Cell-mediated Cytotoxicity and FcγRIIIa Binding. Sci. Rep. 2020, 10, 383. [Google Scholar] [CrossRef]
- Goetze, A.M.; Liu, Y.D.; Zhang, Z.; Shah, B.; Lee, E.; Bondarenko, P.V.; Flynn, G.C. High-mannose glycans on the Fc region of therapeutic IgG antibodies increase serum clearance in humans. Glycobiology 2011, 21, 949–959. [Google Scholar] [CrossRef]
- Chu, G.C.; Chelius, D.; Xiao, G.; Khor, H.K.; Coulibaly, S.; Bondarenko, P.V. Accumulation of succinimide in a recombinant monoclonal antibody in mildly acidic buffers under elevated temperatures. Pharm. Res. 2007, 24, 1145–1156. [Google Scholar] [CrossRef]
- Huang, H.Z.; Nichols, A.; Liu, D. Direct identification and quantification of aspartyl succinimide in an IgG2 mAb by RapiGest assisted digestion. Anal. Chem. 2009, 81, 1686–1692. [Google Scholar] [CrossRef]
- Martin, B.L.; Wu, D.; Tabatabai, L.; Graves, D.J. Formation of cyclic imide-like structures upon the treatment of calmodulin and a calmodulin peptide with heat. Arch. Biochem. Biophys. 1990, 276, 94–101. [Google Scholar] [CrossRef]
- Valliere-Douglass, J.; Jones, L.; Shpektor, D.; Kodama, P.; Wallace, A.; Balland, A.; Bailey, R.; Zhang, Y. Separation and characterization of an IgG2 antibody containing a cyclic imide in CDR1 of light chain by hydrophobic interaction chromatography and mass spectrometry. Anal. Chem. 2008, 80, 3168–3174. [Google Scholar] [CrossRef]
- Xiao, G.; Bondarenko, P.V.; Jacob, J.; Chu, G.C.; Chelius, D. 18O labeling method for identification and quantification of succinimide in proteins. Anal. Chem. 2007, 79, 2714–2721. [Google Scholar] [CrossRef]
- Yan, B.; Steen, S.; Hambly, D.; Valliere-Douglass, J.; Bos, T.V.; Smallwood, S.; Yates, Z.; Arroll, T.; Han, Y.; Gadgil, H.; et al. Succinimide formation at Asn 55 in the complementarity determining region of a recombinant monoclonal antibody IgG1 heavy chain. J. Pharm. Sci. 2009, 98, 3509–3521. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.C.; Joe, K.; Zhang, Y.; Adriano, A.; Wang, Y.; Gazzano-Santoro, H.; Keck, R.G.; Deperalta, G.; Ling, V. Accurate determination of succinimide degradation products using high fidelity trypsin digestion peptide map analysis. Anal. Chem. 2011, 83, 5912–5919. [Google Scholar] [CrossRef] [PubMed]
- Demmon, S.; Bhargava, S.; Ciolek, D.; Halley, J.; Jaya, N.; Joubert, M.K.; Koepf, E.; Smith, P.; Trexler-Schmidt, M.; Tsai, P. A cross-industry forum on benchmarking critical quality attribute identification and linkage to process characterization studies. Biologicals 2020, 67, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Ponniah, G.; Zhang, H.-M.; Nowak, C.; Neill, A.; Gonzalez-Lopez, N.; Patel, R.; Cheng, G.; Kita, A.Z.; Andrien, B. In vitro and in vivo modifications of recombinant and human IgG antibodies. mAbs 2014, 6, 1145–1154. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Valente, J.; Lin, S.; Chennamsetty, N.; Qiu, D.; Bolgar, M. Cyclization of N-Terminal Glutamic Acid to pyro-Glutamic Acid Impacts Monoclonal Antibody Charge Heterogeneity Despite Its Appearance as a Neutral Transformation. J. Pharm. Sci. 2019, 108, 3194–3200. [Google Scholar] [CrossRef]
- Beck, A.; Nowak, C.; Meshulam, D.; Reynolds, K.; Chen, D.; Pacardo, D.B.; Nicholls, S.B.; Carven, G.J.; Gu, Z.; Fang, J.; et al. Risk-Based Control Strategies of Recombinant Monoclonal Antibody Charge Variants. Antibodies 2022, 11, 73. [Google Scholar] [CrossRef] [PubMed]
- Kahle, J.; Zagst, H.; Wiesner, R.; Wätzig, H. Comparative charge-based separation study with various capillary electrophoresis (CE) modes and cation exchange chromatography (CEX) for the analysis of monoclonal antibodies. J. Pharm. Biomed. Anal. 2019, 174, 460–470. [Google Scholar] [CrossRef]
- Zhen, J.; Kim, J.; Zhou, Y.; Gaidamauskas, E.; Subramanian, S.; Feng, P. Antibody characterization using novel ERLIC-MS/MS-based peptide mapping. mAbs 2018, 10, 951–959. [Google Scholar] [CrossRef]
- Pace, A.L.; Wong, R.L.; Zhang, Y.T.; Kao, Y.-H.; Wang, Y.J. Asparagine deamidation dependence on buffer type, pH, and temperature. J. Pharm. Sci. 2013, 102, 1712–1723. [Google Scholar] [CrossRef]
- Kourelis, T.V.; Dasari, S.; Theis, J.D.; Ramirez-Alvarado, M.; Kurtin, P.J.; Gertz, M.A.; Zeldenrust, S.R.; Zenka, R.M.; Dogan, A.; Dispenzieri, A. Clarifying immunoglobulin gene usage in systemic and localized immunoglobulin light-chain amyloidosis by mass spectrometry. Blood 2017, 129, 299–306. [Google Scholar] [CrossRef]
- Sidana, S.; Dasari, S.; Kourelis, T.V.; Dispenzieri, A.; Murray, D.L.; King, R.L.; McPhail, E.D.; Ramirez-Alvarado, M.; Kumar, S.K.; Gertz, M.A. IGVL gene region usage correlates with distinct clinical presentation in IgM vs non-IgM light chain amyloidosis. Blood Adv. 2021, 5, 2101–2105. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Su, Y. Genetic pathogenesis of immunoglobulin light chain amyloidosis: Basic characteristics and clinical applications. Exp. Hematol. Oncol. 2021, 10, 43. [Google Scholar] [CrossRef] [PubMed]
- Trotta, E.; Bessette, P.H.; Silveria, S.L.; Ely, L.K.; Jude, K.M.; Le, D.T.; Holst, C.R.; Coyle, A.; Potempa, M.; Lanier, L.L.; et al. A human anti-IL-2 antibody that potentiates regulatory T cells by a structure-based mechanism. Nat. Med. 2018, 24, 1005–1014. [Google Scholar] [CrossRef] [PubMed]
- Blancas-Mejia, L.M.; Misra, P.; Dick, C.J.; Cooper, S.A.; Redhage, K.R.; Bergman, M.R.; Jordan, T.L.; Maar, K.; Ramirez-Alvarado, M. Immunoglobulin light chain amyloid aggregation. Chem. Commun. 2018, 54, 10664–10674. [Google Scholar] [CrossRef]
- Qin, Z.; Hu, D.; Zhu, M.; Fink, A.L. Structural characterization of the partially folded intermediates of an immunoglobulin light chain leading to amyloid fibrillation and amorphous aggregation. Biochemistry 2007, 46, 3521–3531. [Google Scholar] [CrossRef] [PubMed]
- Schlenzig, D.; Rönicke, R.; Cynis, H.; Ludwig, H.-H.; Scheel, E.; Reymann, K.; Saido, T.; Hause, G.; Schilling, S.; Demuth, H.-U. N-Terminal pyroglutamate formation of Aβ38 and Aβ40 en-forces oligomer formation and potency to disrupt hippocampal long-term potentiation. J. Neurochem. 2012, 121, 774–784. [Google Scholar] [CrossRef] [PubMed]
- Schlenzig, D.; Manhart, S.; Cinar, Y.; Kleinschmidt, M.; Hause, G.; Willbold, D.; Funke, S.A.; Schilling, S.; Demuth, H.-U. Pyroglutamate formation influences solubility and amyloi-dogenicity of amyloid peptides. Biochemistry 2009, 48, 7072–7078. [Google Scholar] [CrossRef]
- Cynis, H.; Frost, J.L.; Crehan, H.; Lemere, C.A. Immunotherapy targeting pyroglutamate-3 Aβ: Prospects and challenges. Mol. Neurodegener. 2016, 11, 48. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Conditions | Acidic % | CEX Main % | Basic % | Acidic % | cIEF Main % | Basic |
|---|---|---|---|---|---|---|
| Control | 19 | 78 | 2.5 | 29 | 66 | 4.4 |
| 25 °C, pH 6.0, 1 month | 25 | 73 | 2.5 | 34 | 63 | 3.6 |
| 40 °C, pH 6.0, 2 weeks | 50 | 34 | 16.1 | 59 | 37 | 3.8 |
| 40 °C, pH 6.0, 1 month | 56 | 13 | 30.6 | 78 | 19.6 | 2.7 |
| Sample Conditions | Percentage of Deamidation | N-Terminal Clipping (%) | ||
|---|---|---|---|---|
| N-1 | N-327 | N386/391 | ||
| A | 78.2 | 8.5 | 5.0 | 1.4 |
| B | 44.4 | 2.6 | 4.7 | 2.9 |
| C | 4.6 | 30.3 | 7.4 | 5.2 |
| D | 1.4 | 11.3 | 11.5 | 8.5 |
| E | 0.4 | 0.8 | 5.8 | 5.8 |
| F | 0.5 | 0.7 | 4.9 | 27.1 |
| Modification | Peptide | Theoretical m/z (z) | Observed m/z | R.T. (min) |
|---|---|---|---|---|
| Unmodified | NFMLTQPHSVSESPGK | 879.9276 (+2) | 879.9237 | 31.4 |
| N1 deamidated | DFMLTQPHSVSESPGK | 880.4196 (+2) | 880.4162 | 32.3 |
| D(iso)FMLTQPHSVSESPGK | 880.4196 (+2) | 880.4163 | 33.0 | |
| N1 succinimide | SucFMLTQPHSVSESPGK | 871.4143 (+2) | 871.4152 | 40.0 |
| M3 oxidation | NFM[O]LTQPHSVSESPGK | 887.9251 (+2) | 887.9222 | 28.7 |
| N 1-clipped | FMLTQPHSVSESPGK | 822.9061 (+2) | 822.9057 | 30.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhen, J.; Lee, J.; Wang, Y.; McLaughlin, L.; Yang, F.; Li, Z.; Wang, J. Characterization of N-Terminal Asparagine Deamidation and Clipping of a Monoclonal Antibody. Antibodies 2023, 12, 59. https://doi.org/10.3390/antib12030059
Zhen J, Lee J, Wang Y, McLaughlin L, Yang F, Li Z, Wang J. Characterization of N-Terminal Asparagine Deamidation and Clipping of a Monoclonal Antibody. Antibodies. 2023; 12(3):59. https://doi.org/10.3390/antib12030059
Chicago/Turabian StyleZhen, Jing, Jennifer Lee, Yueyang Wang, Lena McLaughlin, Fei Yang, Zhengjian Li, and Jihong Wang. 2023. "Characterization of N-Terminal Asparagine Deamidation and Clipping of a Monoclonal Antibody" Antibodies 12, no. 3: 59. https://doi.org/10.3390/antib12030059