
Evaluation of Molecular Simulations and Deep Learning Prediction of Antibodies’ Recognition of TRBC1 and TRBC2

Abstract
:1. Introduction
2. Materials and Methods
2.1. AI Model Training and Prediction of Antibody–Antigen Binding Affinity
2.2. Molecular Dynamics Simulations and Analysis
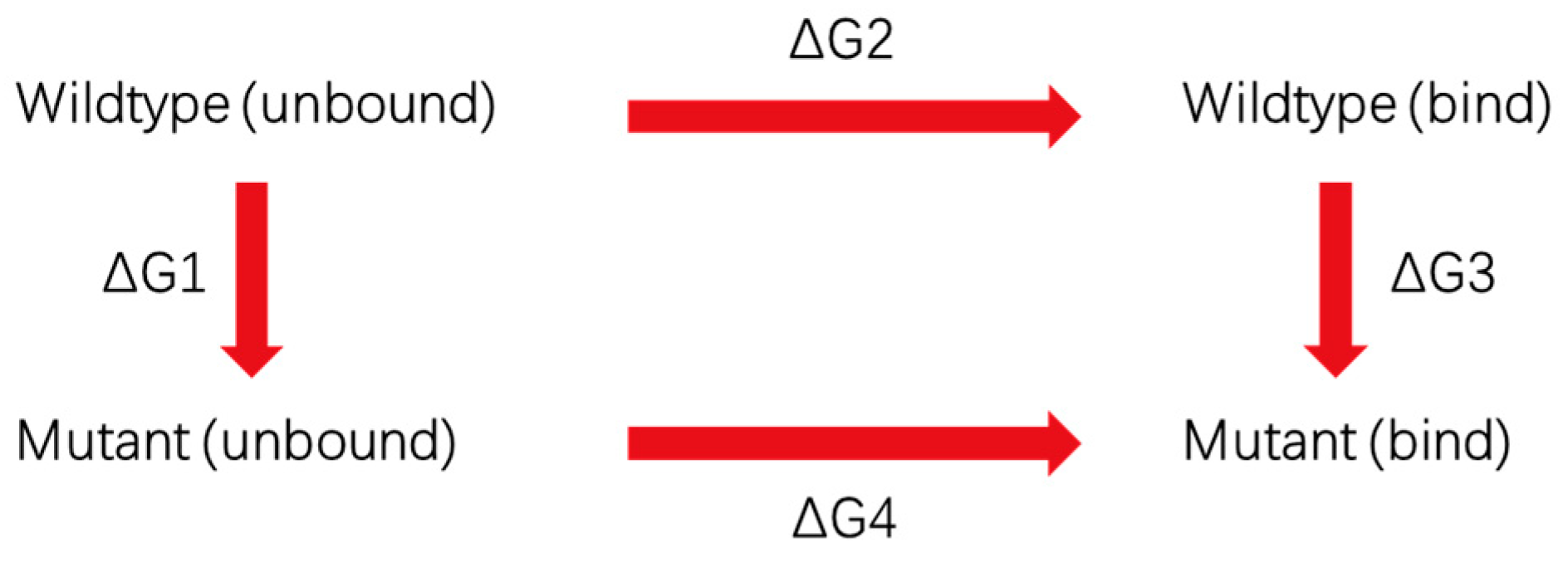
2.3. NAMD-Free Energy Perturbation Protocol
3. Results
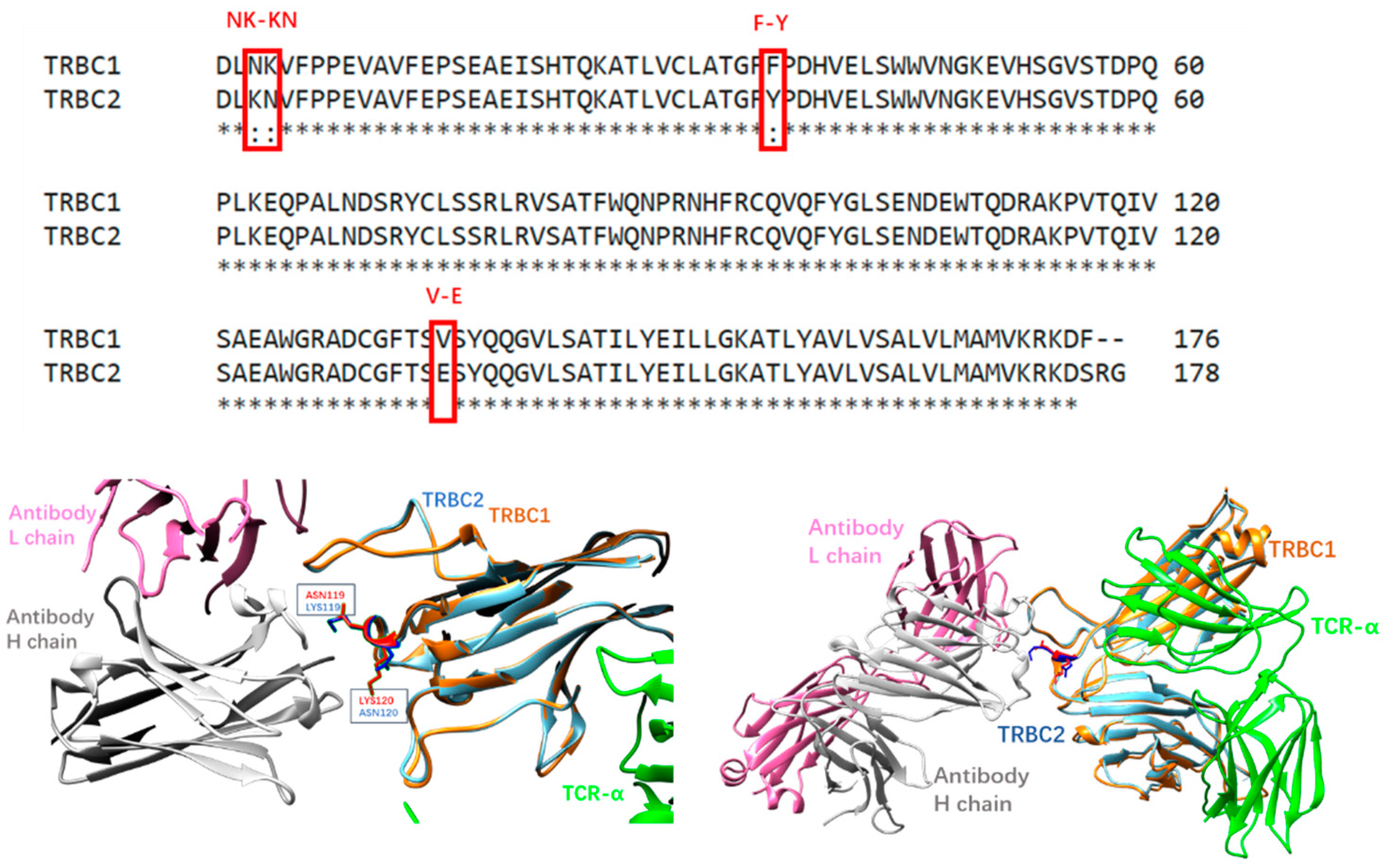
3.1. AI Prediction of Mutation Effects on JOVI-1’s Recognition of TRBC1 and TRBC2
3.2. Prediction of Mutation Effects on JOVI-1’s Recognition of TRBC1 and TRBC2 with Free Energy Perturbation
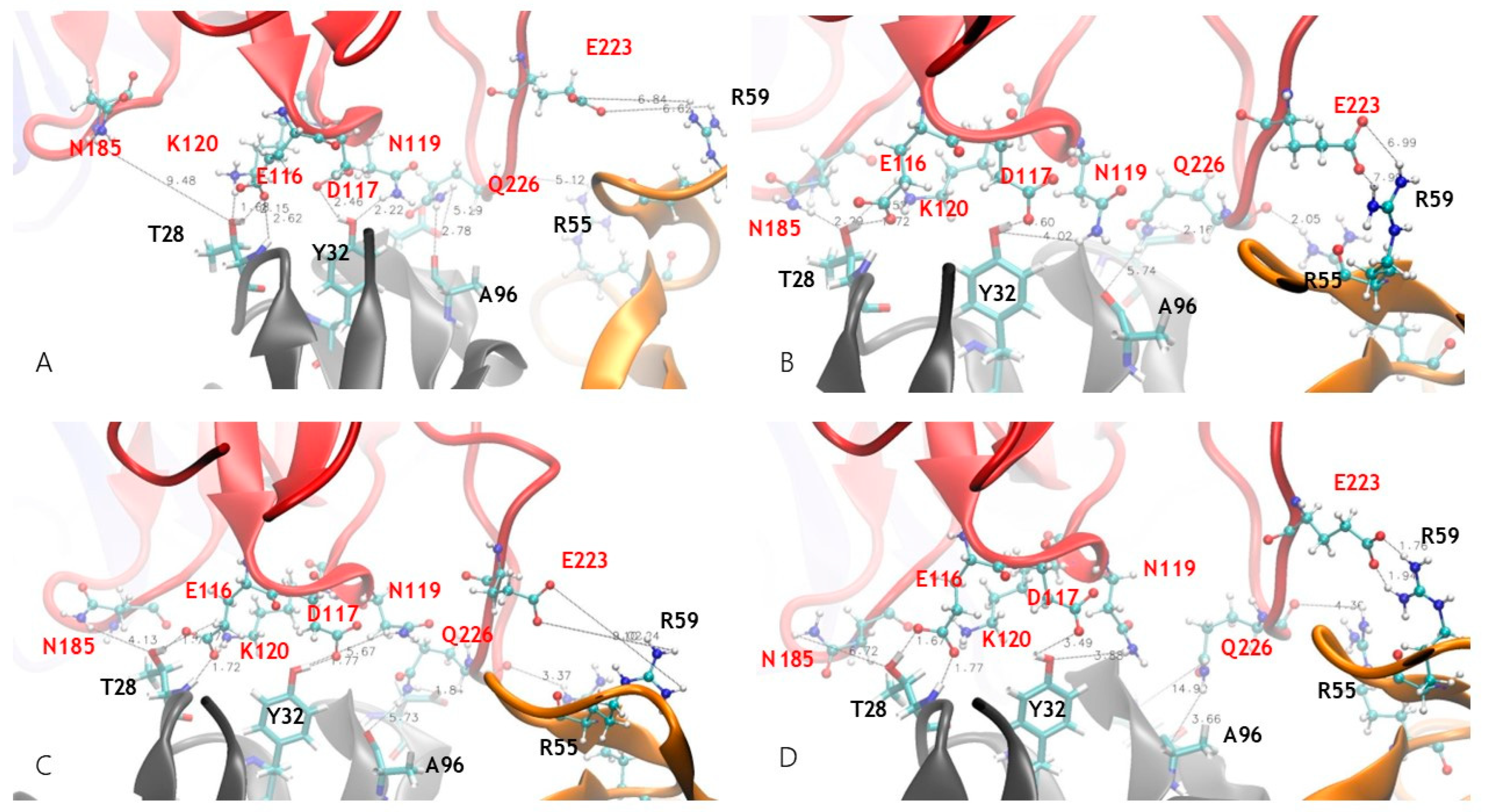
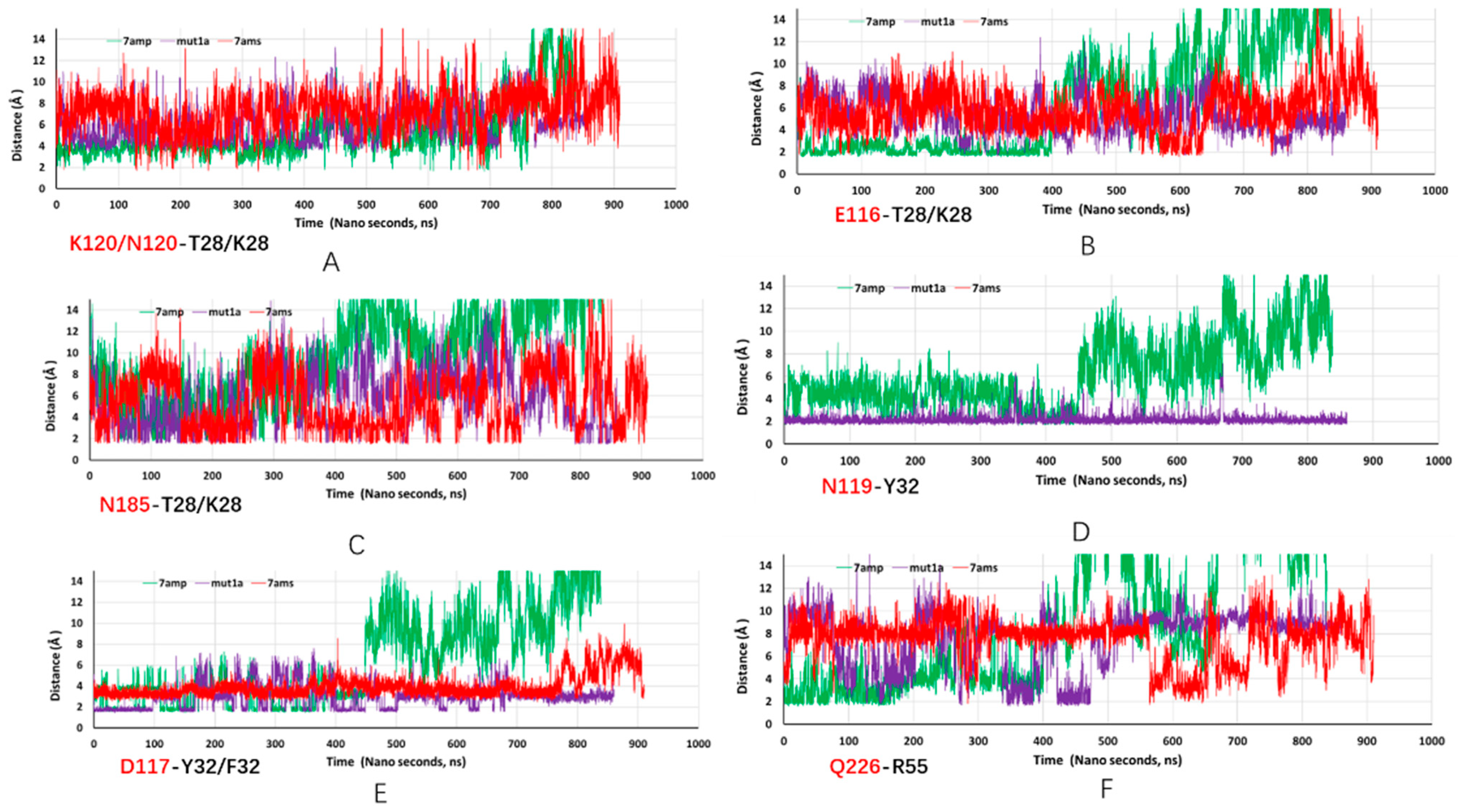
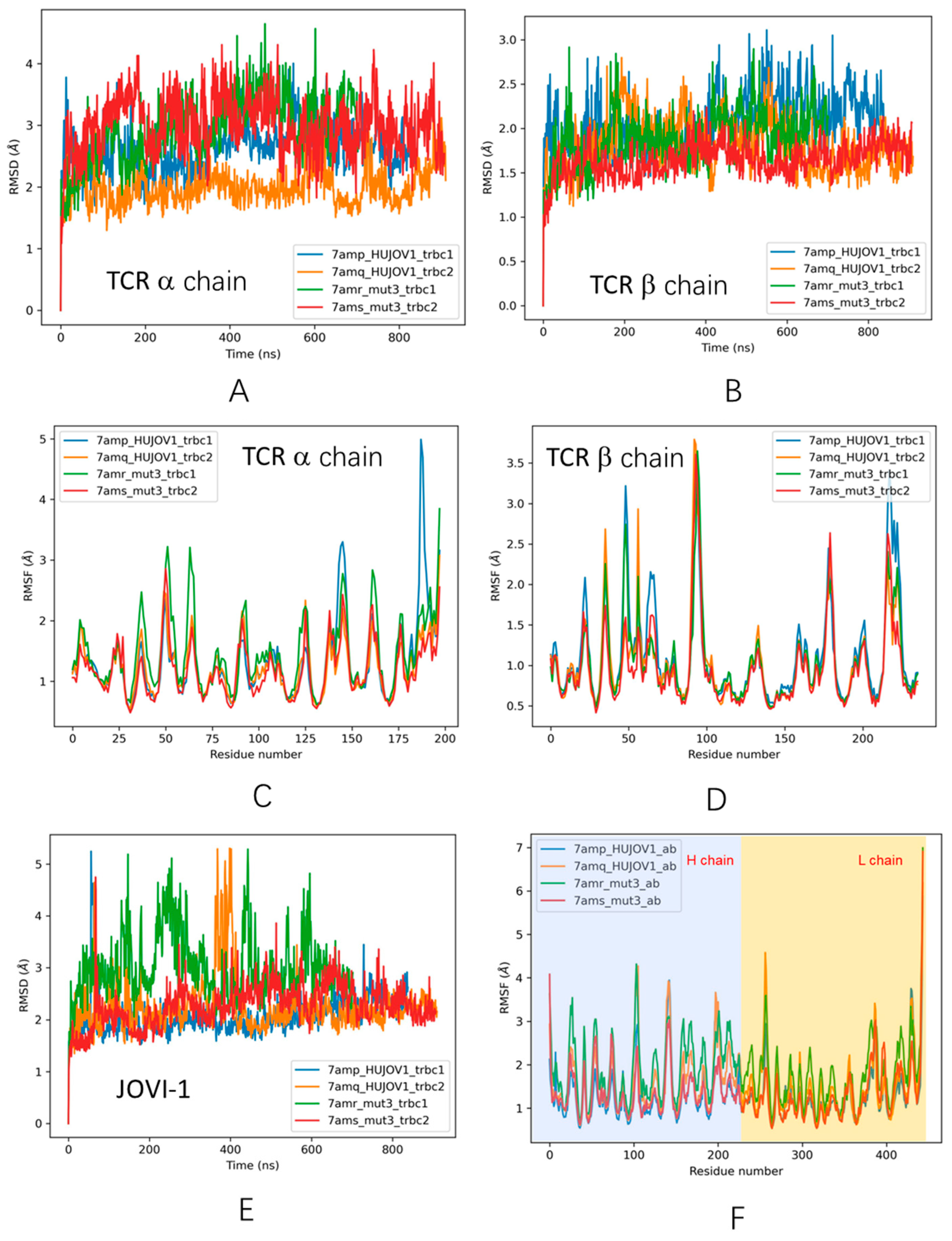
3.3. Dynamic Features of JOVI-1 and Mutants’ Recognition of TRBC1 and TRBC2
4. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Conley, J.M.; Berg, L.J. TCR signaling: It’s all about the numbers. Nat. Immunol. 2019, 20, 1415–1416. [Google Scholar] [CrossRef] [PubMed]
- Viney, J.L.; Prosser, H.M.; Hewitt, C.R.; Lamb, J.R.; Owen, M.J. Generation of monoclonal antibodies against a human T cell receptor beta chain expressed in transgenic mice. Hybridoma 1992, 11, 701–713. [Google Scholar] [CrossRef] [PubMed]
- Maciocia, P.M.; Wawrzyniecka, P.A.; Philip, B.; Ricciardelli, I.; Akarca, A.U.; Onuoha, S.C.; Legut, M.; Cole, D.K.; Sewell, A.K.; Gritti, G.; et al. Targeting the T cell receptor beta-chain constant region for immunotherapy of T cell malignancies. Nat. Med. 2017, 23, 1416–1423. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Garcia, N.; Lima, M.; Villamor, N.; Moran-Plata, F.J.; Barrena, S.; Mateos, S.; Caldas, C.; Balanzategui, A.; Alcoceba, M.; Dominguez, A.; et al. Anti-TRBC1 Antibody-Based Flow Cytometric Detection of T-Cell Clonality: Standardization of Sample Preparation and Diagnostic Implementation. Cancers 2021, 13, 4379. [Google Scholar] [CrossRef]
- Munoz-Garcia, N.; Moran-Plata, F.J.; Villamor, N.; Lima, M.; Barrena, S.; Mateos, S.; Caldas, C.; van Dongen, J.J.M.; Orfao, A.; Almeida, J. High-Sensitive TRBC1-Based Flow Cytometric Assessment of T-Cell Clonality in Talphabeta-Large Granular Lymphocytic Leukemia. Cancers 2022, 14, 408. [Google Scholar] [CrossRef]
- Ma, Y.; Zheng, J.; Li, X.; Song, L.; Liu, W.; Li, J.; Shang, F.; Zhang, Y.; Du, W. Identification of T cell clones by TRBC1. J. Clin. Hematol. 2023, 36, 260–264. [Google Scholar]
- Zhang, C.; Palashati, H.; Rong, Z.; Lin, N.; Shen, L.; Liu, Y.; Li, S.; Yu, B.; Yang, W.; Lu, Z. Pre-depletion of TRBC1+ T cells promotes the therapeutic efficacy of anti-TRBC1 CAR-T for T-cell malignancies. Mol. Cancer 2020, 19, 162. [Google Scholar] [CrossRef]
- Paul, S.; Pearlman, A.H.; Douglass, J.; Mog, B.J.; Hsiue, E.H.; Hwang, M.S.; DiNapoli, S.R.; Konig, M.F.; Brown, P.A.; Wright, K.M.; et al. TCR beta chain-directed bispecific antibodies for the treatment of T cell cancers. Sci. Transl. Med. 2021, 13, eabd3595. [Google Scholar] [CrossRef]
- Ferrari, M.; Baldan, V.; Wawrzyniecka, P.A.; Bulek, A.; Kinna, A.; Ma, B.; Bugda, R.; Akbar, Z.; Srivastava, S.; Ghongane, P.; et al. Structure-Guided Engineering of Immunotherapies Targeting TRBC1 and TRBC2 in T Cell Malignancies; PREPRINT (Version 1); Research Square: Durham, NC, USA, 2022. [Google Scholar]
- Zhao, J.; Nussinov, R.; Wu, W.J.; Ma, B.Y. In Silico Methods in Antibody Design. Antibodies 2018, 7, 15. [Google Scholar] [CrossRef]
- Chowdhury, R.; Allan, M.F.; Maranas, C.D. OptMAVEn-2.0: De novo Design of Variable Antibody Regions against Targeted Antigen Epitopes. Antibodies 2018, 7, 23. [Google Scholar] [CrossRef]
- Zhang, M.Z.; Zheng, J.; Nussinov, R.; Ma, B.Y. Molecular Recognition between A-Specific Single-Domain Antibody and A Misfolded Aggregates. Antibodies 2018, 7, 25. [Google Scholar] [CrossRef]
- Zhao, J.; Mohan, N.; Nussinov, R.; Ma, B.; Wu, W.J. Trastuzumab Blocks the Receiver Function of HER2 Leading to the Population Shifts of HER2-Containing Homodimers and Heterodimers. Antibodies 2021, 10, 7. [Google Scholar] [CrossRef] [PubMed]
- Bai, G.; Ge, Y.; Su, Y.; Chen, S.; Zeng, X.; Lu, H.; Ma, B. Computational Construction of a Single-Chain Bi-Paratopic Antibody Allosterically Inhibiting TCR-Staphylococcal Enterotoxin B Binding. Front. Immunol. 2021, 12, 732938. [Google Scholar] [CrossRef] [PubMed]
- Hummer, A.M.; Abanades, B.; Deane, C.M. Advances in computational structure-based antibody design. Curr. Opin. Struct. Biol. 2022, 74, 102379. [Google Scholar] [CrossRef] [PubMed]
- Raybould, M.I.J.; Marks, C.; Krawczyk, K.; Taddese, B.; Nowak, J.; Lewis, A.P.; Bujotzek, A.; Shi, J.; Deane, C.M. Five computational developability guidelines for therapeutic antibody profiling. Proc. Natl. Acad. Sci. USA 2019, 116, 4025–4030. [Google Scholar] [CrossRef]
- Chen, Y.; Wei, G.; Zhao, J.; Nussinov, R.; Ma, B. Computational Investigation of Gantenerumab and Crenezumab Recognition of Abeta Fibrils in Alzheimer’s Disease Brain Tissue. ACS Chem. Neurosci. 2020, 11, 3233–3244. [Google Scholar] [CrossRef]
- Zhao, J.; Nussinov, R.; Ma, B.Y. Antigen binding allosterically promotes Fc receptor recognition. mAbs 2019, 11, 58–74. [Google Scholar] [CrossRef]
- Saetang, J.; Sangkhathat, S.; Jangphattananont, N.; Khopanlert, W.; Julamanee, J.; Tipmanee, V. Computational discovery of binding mode of anti-TRBC1 antibody and predicted key amino acids of TRBC1. Sci. Rep. 2022, 12, 1760. [Google Scholar] [CrossRef]
- Wong, W.K.; Robinson, S.A.; Bujotzek, A.; Georges, G.; Lewis, A.P.; Shi, J.; Snowden, J.; Taddese, B.; Deane, C.M. Ab-Ligity: Identifying sequence-dissimilar antibodies that bind to the same epitope. mAbs 2021, 13, 1873478. [Google Scholar] [CrossRef]
- Dunbar, J.; Krawczyk, K.; Leem, J.; Marks, C.; Nowak, J.; Regep, C.; Georges, G.; Kelm, S.; Popovic, B.; Deane, C.M. SAbPred: A structure-based antibody prediction server. Nucleic Acids Res. 2016, 44, W474–W478. [Google Scholar] [CrossRef]
- Graves, J.; Byerly, J.; Priego, E.; Makkapati, N.; Parish, S.V.; Medellin, B.; Berrondo, M. A Review of Deep Learning Methods for Antibodies. Antibodies 2020, 9, 12. [Google Scholar] [CrossRef]
- Ruffolo, J.A.; Sulam, J.; Gray, J.J. Antibody structure prediction using interpretable deep learning. Patterns 2022, 3, 100406. [Google Scholar] [CrossRef]
- Schneider, C.; Buchanan, A.; Taddese, B.; Deane, C.M. DLAB: Deep learning methods for structure-based virtual screening of antibodies. Bioinformatics 2022, 38, 377–383. [Google Scholar] [CrossRef]
- Bai, G.; Sun, C.; Guo, Z.; Wang, Y.; Zeng, X.; Su, Y.; Zhao, Q.; Ma, B. Accelerating antibody discovery and design with artificial intelligence: Recent. advances and prospects. Semin. Cancer Biol. 2023, 95, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Hie, B.L.; Shanker, V.R.; Xu, D.; Bruun, T.U.J.; Weidenbacher, P.A.; Tang, S.; Wu, W.; Pak, J.E.; Kim, P.S. Efficient evolution of human antibodies from general protein language models. Nat. Biotechnol. 2023. [Google Scholar] [CrossRef] [PubMed]
- Jankauskaite, J.; Jimenez-Garcia, B.; Dapkunas, J.; Fernandez-Recio, J.; Moal, I.H. SKEMPI 2.0: An updated benchmark of changes in protein-protein binding energy, kinetics and thermodynamics upon mutation. Bioinformatics 2019, 35, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Sirin, S.; Apgar, J.R.; Bennett, E.M.; Keating, A.E. AB-Bind: Antibody binding mutational database for computational affinity predictions. Protein Sci. 2016, 25, 393–409. [Google Scholar] [CrossRef]
- Ruffolo, J.A.; Gray, J.J.; Sulam, J. Deciphering antibody affinity maturation with language models and weakly supervised learning. arXiv 2021, arXiv:2112.07782. [Google Scholar]
- Brandes, N.; Ofer, D.; Peleg, Y.; Rappoport, N.; Linial, M. ProteinBERT: A universal deep-learning model of protein sequence and function. Bioinformatics 2022, 38, 2102–2110. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E., III; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed]
- MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef] [PubMed]
- Ichiye, T.; Karplus, M. Collective motions in proteins: A covariance analysis of atomic fluctuations in molecular dynamics and normal mode simulations. Proteins Struct. Funct. Bioinform. 1991, 11, 205–217. [Google Scholar] [CrossRef]
- Glykos, N.M. Software news and updates carma: A molecular dynamics analysis program. J. Comput. Chem. 2006, 27, 1765–1768. [Google Scholar] [CrossRef] [PubMed]
- Kale, L.; Skeel, R.; Bhandarkar, M.; Brunner, R.; Gursoy, A.; Krawetz, N.; Phillips, J.; Shinozaki, A.; Varadarajan, K.; Schulten, K. NAMD2: Greater scalability for parallel molecular dynamics. J. Comput. Phys. 1999, 151, 283–312. [Google Scholar] [CrossRef]
- Rangarajan, S.; He, Y.A.; Chen, Y.H.; Kerzic, M.C.; Ma, B.Y.; Gowthaman, R.; Pierce, B.G.; Nussinov, R.; Mariuzza, R.A.; Orban, J. Peptide-MHC (pMHC) binding to a human antiviral T cell receptor induces long-range allosteric communication between pMHC- and CD3-binding sites. J. Biol. Chem. 2018, 293, 15991–16005. [Google Scholar] [CrossRef]
- He, Y.; Agnihotri, P.; Rangarajan, S.; Chen, Y.; Kerzic, M.C.; Ma, B.; Nussinov, R.; Mariuzza, R.A.; Orban, J. Peptide-MHC Binding Reveals Conserved Allosteric Sites in MHC Class I- and Class II-Restricted T Cell Receptors (TCRs). J. Mol. Biol. 2020, 432, 166697. [Google Scholar] [CrossRef]
- Mason, D.M.; Friedensohn, S.; Weber, C.R.; Jordi, C.; Wagner, B.; Meng, S.M.; Ehling, R.A.; Bonati, L.; Dahinden, J.; Gainza, P.; et al. Optimization of therapeutic antibodies by predicting antigen specificity from antibody sequence via deep learning. Nat. Biomed. Eng. 2021, 5, 600–612. [Google Scholar] [CrossRef]
- Makowski, E.K.; Kinnunen, P.C.; Huang, J.; Wu, L.; Smith, M.D.; Wang, T.; Desai, A.A.; Streu, C.N.; Zhang, Y.; Zupancic, J.M.; et al. Co-optimization of therapeutic antibody affinity and specificity using machine learning models that generalize to novel mutational space. Nat. Commun. 2022, 13, 3788. [Google Scholar] [CrossRef]
- Kim, J.; McFee, M.; Fang, Q.; Abdin, O.; Kim, P.M. Computational and artificial intelligence-based methods for antibody development. Trends Pharmacol. Sci. 2023, 44, 175–189. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
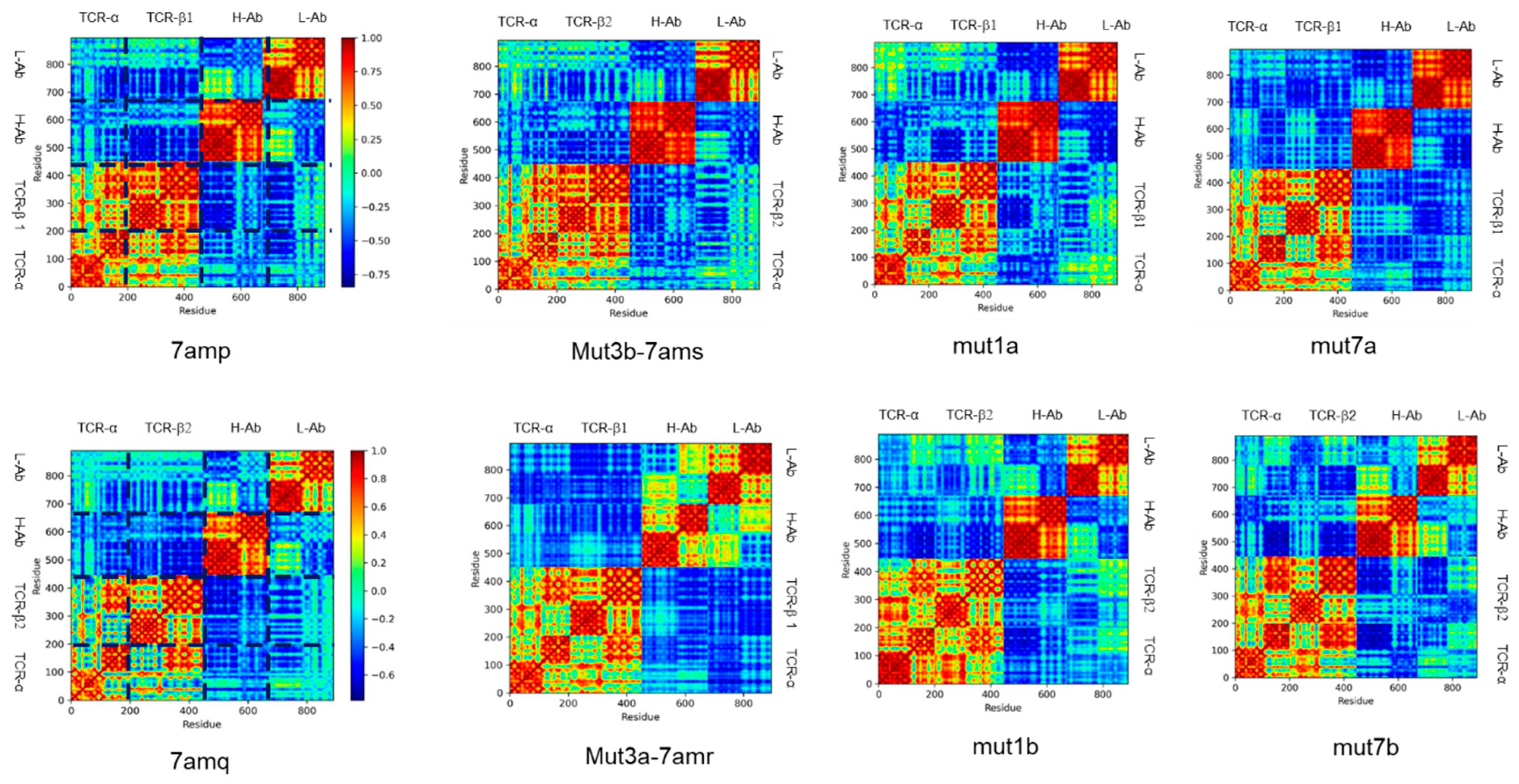{kind=link}
{kind=link}
| Method | Pearson | Spearman |
|---|---|---|
| bASA | 0.22 | |
| DFIRE | 0.31 | |
| dDFIRE | 0.19 | |
| Rosetta | 0.16 | |
| STATIUM | 0.32 | |
| FoldX | 0.34 | |
| Discovery Studio | 0.45 | |
| Our Digiwiser Model | 0.74–0.89 | 0.72–0.78 |
| System | PDB Code | Antibody | Antigen | Affinity | Atoms | Stable Time 1 | |
|---|---|---|---|---|---|---|---|
| Close | Far | ||||||
| wt | 7amp | HuJovi-1 Fab | TRBC1 | −11.8 | 344,304 | >850 ns | >850 ns |
| Wt2 | 7amq | HuJovi-1 Fab | TRBC2 | −6.7 | 325,267 | 530 ns | 550 ns |
| Mut1a | 7amp/T28K | HuJovi-1/T28K | TRBC1 | −11.2 | 344,248 | >860 ns | >860 ns |
| Mut1b | 7amq/T28K | HuJovi-1/T28K | TRBC2 | −7.0 | 325,262 | 65 ns | 250 ns |
| Mut2a | 7amr/N96A | HuJovi-1/T28K/Y32F | TRBC1 | −8.8 | 325,251 | >900 ns | >900 ns |
| Mut2b | 7ams/N96A | HuJovi-1/T28K/Y32F | TRBC2 | −8.2 | 325,180 | >900 ns | >900 ns |
| Mut3a | 7amr | HuJovi-1/KFN | TRBC1 | −7.8 | 325,394 | 125 ns | 150 ns |
| Mut3b | 7ams | HuJovi-1/KFN | TRBC2 | −8.7 | 325,428 | >900 ns | >900 ns |
| Mut4a | 7amr/K28R | HuJovi-1/RFN | TRBC1 | −8.0 | 325,606 | >900 ns | >900 ns |
| Mut4b | 7ams/K28R | HuJovi-1/RFN | TRBC2 | −8.6 | 325,289 | >900 ns | >900 ns |
| Mut5a | 7amp/Y32F | HuJovi-1/Y32F | TRBC1 | −8.7 | 344,366 | >900 ns | >900 ns |
| Mut5b | 7amq/Y32F | HuJovi-1/Y32F | TRBC2 | - | 325,344 | >900 ns | >900 ns |
| Mut6a | 7amp/A96N | HuJovi-1/A96N | TRBC1 | −10.8 | 344,212 | 320 ns | >900 ns |
| Mut6b | 7amq/A96N | HuJovi-1/A96N | TRBC2 | −8.8 | 325,193 | 550 ns | 600 ns |
| Mut7a | 7amr/F32Y | HuJovi-1/T28K/A96N | TRBC1 | −10.2 | 344,405 | 350 ns | >900 ns |
| Mut7b | 7ams/F32Y | HuJovi-1/T28K/A96N | TRBC2 | −8.8 | 325,311 | 30 ns | >900 ns |
| System | Antigen | Affinity | Rank 1 | System | Antigen | Affinity | Rank 2 |
|---|---|---|---|---|---|---|---|
| Mut1a | TRBC1 | −11.2 | 0.408 | Mut1b | TRBC2 | −7.0 | 0.408 |
| Mut2a | TRBC1 | −8.8 | 0.708 | Mut2b | TRBC2 | −8.2 | 0.713 |
| Mut4a | TRBC1 | −8.0 | 0.394 | Mut4b | TRBC2 | −8.6 | 0.404 |
| Mut5a | TRBC2 | −8.7 | 0.532 | ||||
| Mut6a | TRBC1 | −10.8 | 0.144 | Mut6b | TRBC2 | −8.8 | 0.139 |
| Mut7a | TRBC1 | −10.2 | 0.396 | Mut7b | TRBC2 | −8.8 | 0.398 |
| Pearson | 0.543 | Pearson | 0.272 | ||||
| Spearman | 0.143 | Spearman | 0.899 |
| Mut | System | Antigen | ΔΔGexp | ΔΔGfep | |err| | Mut | System | Antigen | ΔΔGexp | ΔΔGfep | |err| |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 7AMP:T28K | Mut1a | TRBC1 | 0.56 | 0.47 | 0.1 | 7AMQ:T28K | Mut1b | TRBC2 | −0.33 | −2.19 | 1.85 |
| 7AMR(N96A) | Mut2a | TRBC1 | −0.97 | −3.19 | 2.22 | 7AMS:N96A | Mut2b | TRBC2 | 0.44 | 0.11 | 0.33 |
| 7AMR:K28R | Mut4a | TRBC1 | −0.19 | −0.32 | 0.13 | 7AMS:K28R | Mut4b | TRBC2 | 0.11 | −2.47 | 2.58 |
| 7AMP:Y32F | Mut5a | TRBC1 | 3.04 | 2.24 | 0.80 | 7AMQ:Y32F | Mut5b | TRBC2 | NB | 10.27 | |
| 7AMP:A96N | Mut6a | TRBC1 | 0.99 | 0.27 | 0.72 | 7AMQ:A96N | Mut6b | TRBC2 | −2.11 | −0.10 | 2.01 |
| 7AMR:F32Y | Mut7a | TRBC1 | −2.38 | 1.35 | 3.72 | 7AMS:F32Y | Mut7b | TRBC2 | −3.78 | −0.12 | 3.66 |
| Average err | 1.28 | 2.09 | |||||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeng, X.; Wang, T.; Kang, Y.; Bai, G.; Ma, B. Evaluation of Molecular Simulations and Deep Learning Prediction of Antibodies’ Recognition of TRBC1 and TRBC2. Antibodies 2023, 12, 58. https://doi.org/10.3390/antib12030058
Zeng X, Wang T, Kang Y, Bai G, Ma B. Evaluation of Molecular Simulations and Deep Learning Prediction of Antibodies’ Recognition of TRBC1 and TRBC2. Antibodies. 2023; 12(3):58. https://doi.org/10.3390/antib12030058
Chicago/Turabian StyleZeng, Xincheng, Tianqun Wang, Yue Kang, Ganggang Bai, and Buyong Ma. 2023. "Evaluation of Molecular Simulations and Deep Learning Prediction of Antibodies’ Recognition of TRBC1 and TRBC2" Antibodies 12, no. 3: 58. https://doi.org/10.3390/antib12030058