
Genome-Wide DNA Methylation Analysis and Functional Validation of Litter Size Traits in Jining Grey Goats
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
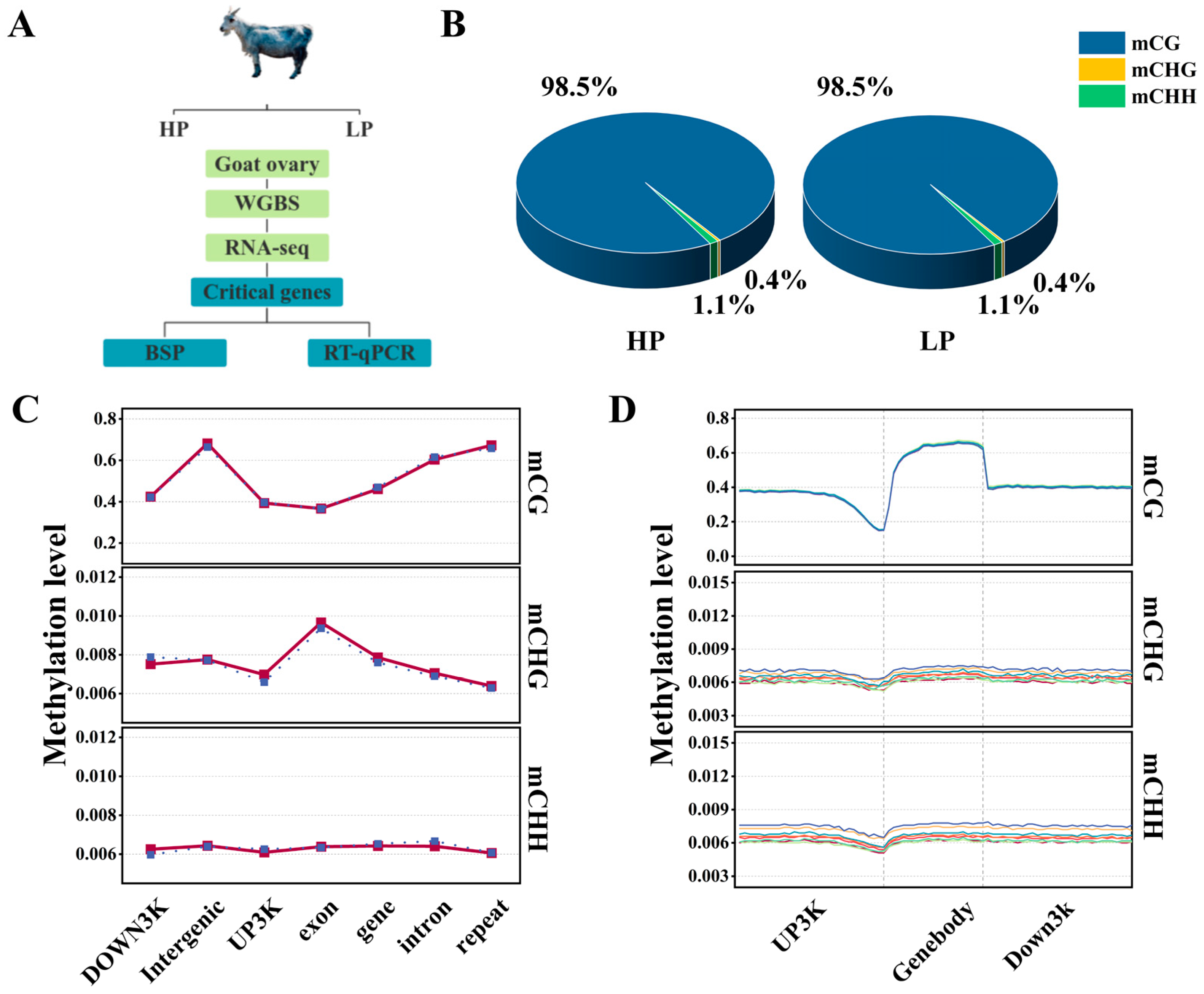
2.1. Animals and Sample Collection
2.2. DNA and RNA Extractions
2.3. Library Preparation
2.4. WGBS and Identification of DMRs
2.5. DMR-Associated Genes (DMGs) and Functional Enrichment Analysis
2.6. RT-qPCR Analysis
2.7. Bisulfite Sequencing PCR (BSP)
2.8. Statistical Analysis
3. Results
3.1. Genome-Wide Methylation Mapping
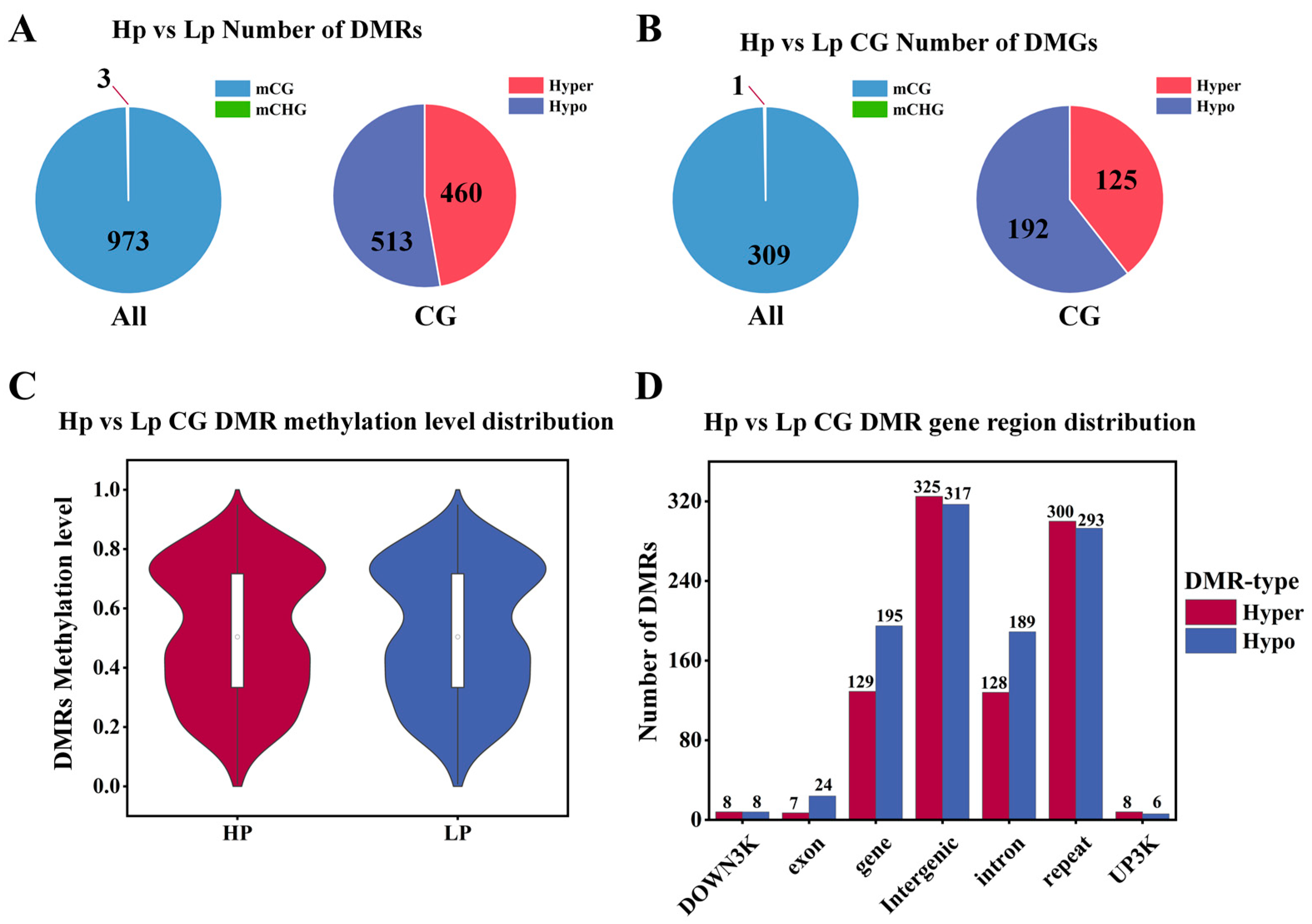
3.2. Differential Methylation Region (DMR) Identification
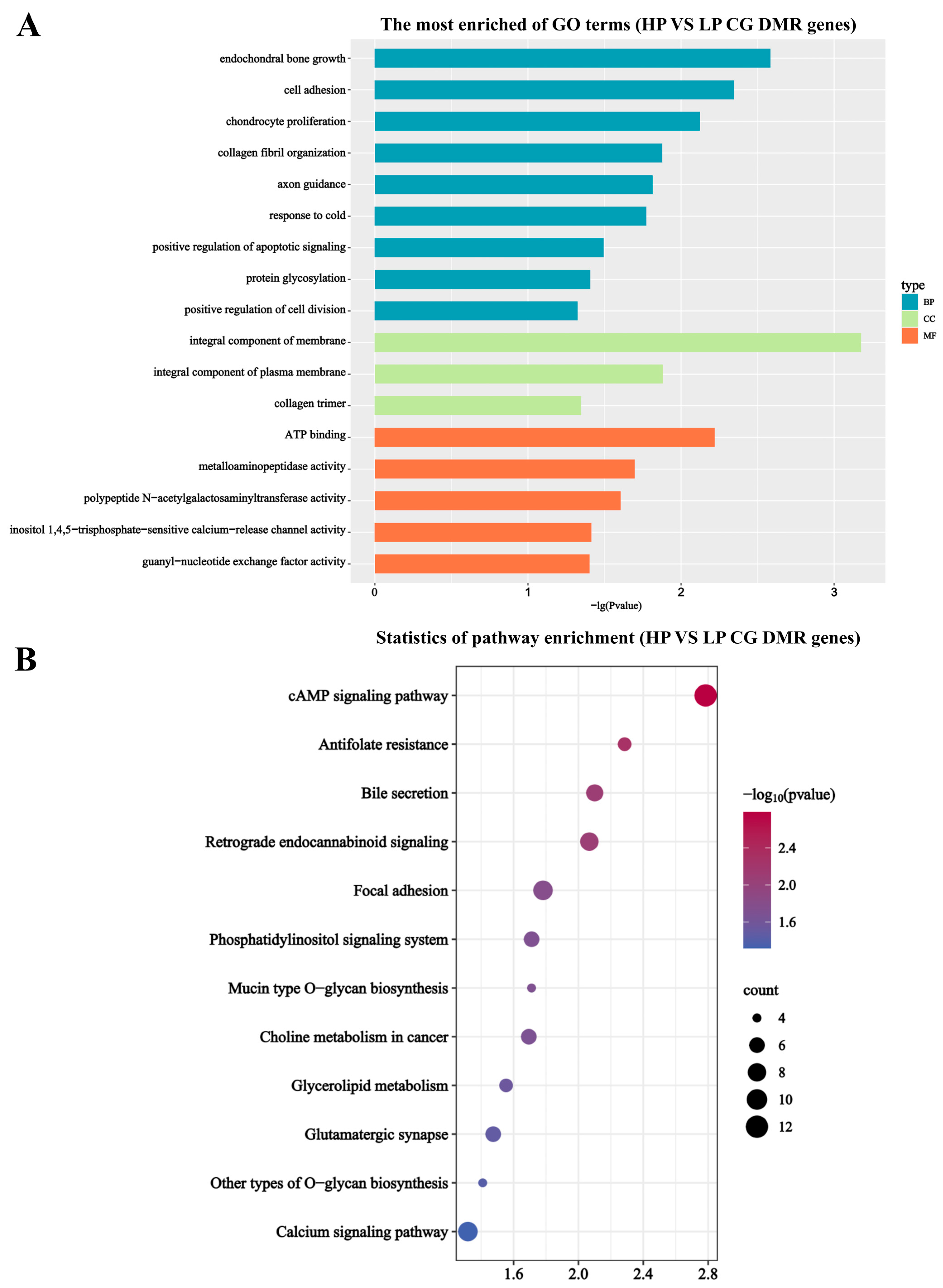
3.3. DMG Functional Enrichment Analysis
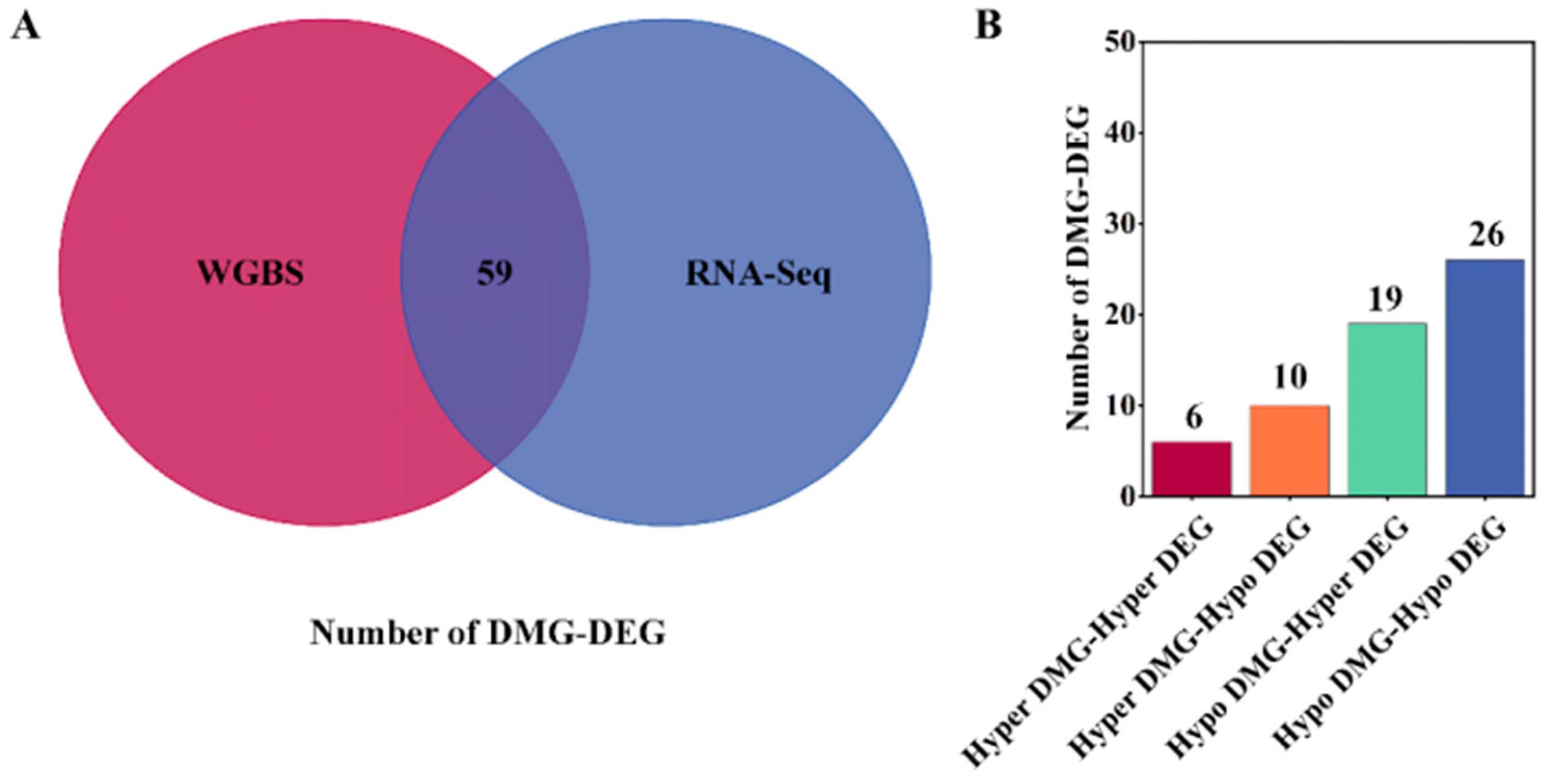
3.4. Screening for DMG Associated with Litter Size Traits
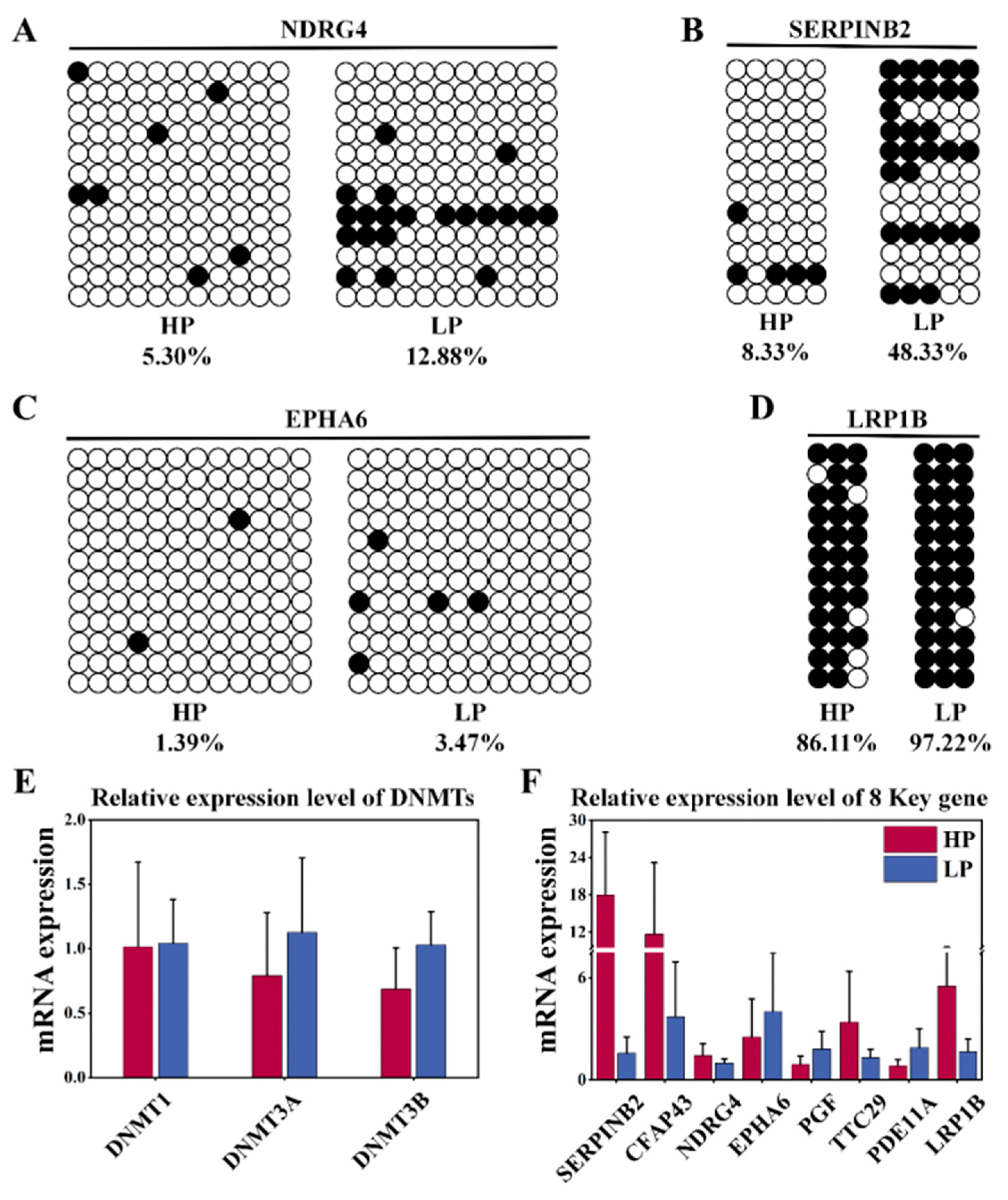
3.5. Key Candidate Gene Validation
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, J.-J.; Zhang, T.; Chen, Q.-M.; Zhang, R.-Q.; Li, L.; Cheng, S.-F.; Shen, W.; Lei, C.-Z. Genomic Signatures of Selection Associated with Litter Size Trait in Jining Gray Goat. Front. Genet. 2020, 11, 286. [Google Scholar] [CrossRef]
- Yao, X.; Li, F.; Wei, Z.; Ei-Samahy, M.A.; Feng, X.; Yang, F.; Wang, F. Integrative Genome-Wide DNA Methylome and Transcriptome Analysis of Ovaries from Hu Sheep with High and Low Prolific. Front. Cell Dev. Biol. 2022, 10, 820558. [Google Scholar] [CrossRef]
- Liu, A.; Liu, M.; Li, Y.; Chen, X.; Zhang, L.; Tian, S. Differential expression and prediction of function of lncRNAs in the ovaries of low and high fecundity Hanper sheep. Reprod. Domest. Anim. 2021, 56, 604–620. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, R.; Pan, C.; Chen, H.; Qu, L.; Wu, L.; Guo, Z.; Zhu, H.; Lan, X. Genetic Variations and mRNA Expression of Goat DNAH1 and Their Associations with Litter Size. Cells 2022, 11, 1371. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Wang, Z.; Wang, X.; Li, Y.; Qu, L.; Lan, X. A novel 4-bp insertion within the goat CFAP43 gene and its association with litter size. Small Rumin. Res. 2021, 202, 106456. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, X.; Ruan, Y.; Guo, W.; Chen, J.; Tang, W.; Ji, Q.; Fu, K. Effect of CTSS non-synonymous mutations on litter size in Qianbei Ma goats. Front. Vet. Sci. 2023, 10, 1276673. [Google Scholar] [CrossRef] [PubMed]
- Smołucha, G.; Gurgul, A.; Jasielczuk, I.; Kawęcka, A.; Miksza-Cybulska, A. A genome-wide association study for prolificacy in three Polish sheep breeds. J. Appl. Genet. 2021, 62, 323–326. [Google Scholar] [CrossRef]
- Zhu, H.; Zhang, Y.; Bai, Y.; Yang, H.; Yan, H.; Liu, J.; Shi, L.; Song, X.; Li, L.; Dong, S.; et al. Relationship between SNPs of POU1F1 Gene and Litter Size and Growth Traits in Shaanbei White Cashmere Goats. Animals 2019, 9, 114. [Google Scholar] [CrossRef] [PubMed]
- Kang, B.; Wang, J.; Zhang, H.; Shen, W.; El-Mahdy Othman, O.; Zhao, Y.; Min, L. Genome-wide profile in DNA methylation in goat ovaries of two different litter size populations. J. Anim. Physiol. Anim. Nutr. 2021, 106, 239–249. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, F.; Feng, X.; Yang, H.; Zhu, A.; Pang, J.; Han, L.; Zhang, T.; Yao, X.; Wang, F. Genome-wide analysis of DNA Methylation profiles on sheep ovaries associated with prolificacy using whole-genome Bisulfite sequencing. BMC Genom. 2017, 18, 759. [Google Scholar] [CrossRef]
- Zhou, X.; Yang, S.; Yan, F.; He, K.; Zhao, A. Genome-wide DNA methylation profiles of porcine ovaries in estrus and proestrus. Physiol. Genom. 2018, 50, 714–723. [Google Scholar] [CrossRef]
- Frattini, S.; Capra, E.; Lazzari, B.; McKay, S.D.; Coizet, B.; Talenti, A.; Groppetti, D.; Riccaboni, P.; Pecile, A.; Chessa, S.; et al. Genome-wide analysis of DNA methylation in hypothalamus and ovary of Capra hircus. BMC Genom. 2017, 18, 476. [Google Scholar] [CrossRef]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.-M.; et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Xiang, H.; Zhu, J.; Chen, Q.; Dai, F.; Li, X.; Li, M.; Zhang, H.; Zhang, G.; Li, D.; Dong, Y.; et al. Single base–resolution methylome of the silkworm reveals a sparse epigenomic map. Nat. Biotechnol. 2010, 28, 516–520. [Google Scholar] [CrossRef]
- Catoni, M.; Tsang, J.M.; Greco, A.P.; Zabet, N.R. DMRcaller: A versatile R/Bioconductor package for detection and visualization of differentially methylated regions in CpG and non-CpG contexts. Nucleic Acids Res. 2018, 46, e114. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Mi, F.; Wu, X.; Wang, Z.; Wang, R.; Lan, X. Relationships between the Mini-InDel Variants within the Goat CFAP43 Gene and Body Traits. Animals 2022, 12, 3447. [Google Scholar] [CrossRef] [PubMed]
- Coutton, C.; Vargas, A.S.; Amiri-Yekta, A.; Kherraf, Z.-E.; Ben Mustapha, S.F.; Le Tanno, P.; Wambergue-Legrand, C.; Karaouzène, T.; Martinez, G.; Crouzy, S.; et al. Mutations in CFAP43 and CFAP44 cause male infertility and flagellum defects in Trypanosoma and human. Nat. Commun. 2018, 9, 686. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; He, X.Y.; Wang, F.Y.; Pan, L.X.; Wang, X.Y.; Gan, S.Q.; Di, R.; Chu, M.X. Identification of genes associated with litter size combining genomic approaches in Luzhong mutton sheep. Anim. Genet. 2021, 52, 545–549. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, F.; Gao, G.; Yan, X.; Liu, H.; Liu, Z.; Wang, Z.; He, L.; Lv, Q.; Wang, Z.; et al. Genome-Wide Association Study of Body Weight Traits in Inner Mongolia Cashmere Goats. Front. Vet. Sci. 2021, 8, 752746. [Google Scholar] [CrossRef]
- Qu, X.; Guo, S.; Yan, L.; Zhu, H.; Li, H.; Shi, Z. TNFα-Erk1/2 signaling pathway-regulated SerpinE1 and SerpinB2 are involved in lipopolysaccharide-induced porcine granulosa cell proliferation. Cell. Signal. 2020, 73, 109702. [Google Scholar] [CrossRef]
- Wu, W.B.; Xu, Y.Y.; Cheng, W.W.; Yuan, B.; Zhao, J.R.; Wang, Y.L.; Zhang, H.J. Decreased PGF May Contribute to Trophoblast Dysfunction in Fetal Growth Restriction. Reproduction 2017, 154, 319–329. [Google Scholar] [CrossRef]
- Vrachnis, N.; Kalampokas, E.; Sifakis, S.; Vitoratos, N.; Kalampokas, T.; Botsis, D.; Iliodromiti, Z. Placental growth factor (PlGF): A key to optimizing fetal growth. J. Matern.-Fetal Neonatal Med. 2013, 26, 995–1002. [Google Scholar] [CrossRef]
- Seidenspinner, T.; Tetens, J.; Habier, D.; Bennewitz, J.; Thaller, G. The placental growth factor (PGF)—A positional and functional candidate gene influencing calving ease and stillbirth in German dairy cattle. Anim. Genet. 2011, 42, 22–27. [Google Scholar] [CrossRef]
- Wang, H.; Yang, Q.; Gu, Y.; Zhang, X.; Wang, J.-M.; He, Y.-P.; Shi, Y.; Sun, Z.-G.; Shi, H.-J.; Wang, J. Uterine Expression of NDRG4 Is Induced by Estrogen and Up-Regulated during Embryo Implantation Process in Mice. PLoS ONE 2016, 11, e0155491. [Google Scholar] [CrossRef]
- Jandrey, E.H.F.; Moura, R.P.; Andrade, L.N.S.; Machado, C.L.; Campesato, L.F.; Leite, K.R.M.; Inoue, L.T.; Asprino, P.F.; da Silva, A.P.M.; de Barros, A.C.S.D.; et al. NDRG4 promoter hypermethylation is a mechanistic biomarker associated with metastatic progression in breast cancer patients. npj Breast Cancer 2019, 5, 11. [Google Scholar] [CrossRef] [PubMed]
- Faja, F.; Finocchi, F.; Carlini, T.; Rizzo, F.; Pallotti, F.; Spaziani, M.; Balercia, G.; Lenzi, A.; Paoli, D.; Lombardo, F. PDE11A gene polymorphism in testicular cancer: Sperm parameters and hormonal profile. J. Endocrinol. Investig. 2021, 44, 2273–2284. [Google Scholar] [CrossRef] [PubMed]
- Pathak, A.; Stewart, D.R.; Faucz, F.R.; Xekouki, P.; Bass, S.; Vogt, A.; Zhang, X.; Boland, J.; Yeager, M.; Loud, J.T.; et al. Rare inactivating PDE11A variants associated with testicular germ cell tumors. Endocr. Relat. Cancer 2015, 22, 909–917. [Google Scholar] [CrossRef] [PubMed]
- Laqqan, M.M.; Yassin, M.M. Influence of tobacco cigarette heavy smoking on DNA methylation patterns and transcription levels of MAPK8IP3, GAA, ANXA2, PRRC2A, and PDE11A genes in human spermatozoa. Middle East Fertil. Soc. J. 2021, 26, 41. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, S.; Xu, Y.; Ma, Y.; Zhang, D.; Li, X.; Zhao, S. The DNA Methylation Status of Wnt and Tgfβ Signals Is a Key Factor on Functional Regulation of Skeletal Muscle Satellite Cell Development. Front. Genet. 2019, 10, 220. [Google Scholar] [CrossRef]
- Yuan, X.-L.; Zhang, Z.; Li, B.; Gao, N.; Zhang, H.; Sangild, P.T.; Li, J.-Q. Genome-wide DNA methylation analysis of the porcine hypothalamus-pituitary-ovary axis. Sci. Rep. 2017, 7, 4277. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Liang, Y.; Deng, K.; Zhang, Z.; Zhang, G.; Zhang, Y.; Wang, F. Analysis of DNA methylation profiles during sheep skeletal muscle development using whole-genome bisulfite sequencing. BMC Genom. 2020, 21, 327. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Cui, Y.; Gu, X. Genome-wide DNA methylation profiles changes associated with constant heat stress in pigs as measured by bisulfite sequencing. Sci. Rep. 2016, 6, 27507. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Pelizzola, M.; Kida, Y.S.; Hawkins, R.D.; Nery, J.R.; Hon, G.; Antosiewicz-Bourget, J.; O’Malley, R.; Castanon, R.; Klugman, S.; et al. Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature 2011, 471, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.-L.; Gao, N.; Xing, Y.; Zhang, H.-B.; Zhang, A.-L.; Liu, J.; He, J.-L.; Xu, Y.; Lin, W.-M.; Chen, Z.-M.; et al. Profiling the genome-wide DNA methylation pattern of porcine ovaries using reduced representation bisulfite sequencing. Sci. Rep. 2016, 6, 22138. [Google Scholar] [CrossRef] [PubMed]
- Bourc’his, D.; Malik, H.S.; Molaro, A.; Satta, Y. Dynamic Evolution of De Novo DNA Methyltransferases in Rodent and Primate Genomes. Mol. Biol. Evol. 2020, 37, 1882–1892. [Google Scholar] [CrossRef]
- Stevenson, T.J.; Bowman, A.S.; Campbell, E.M.; Lorgen, M.; Coyle, C.S.; Lynch, E.W.J. Cyclical DNA Methyltransferase 3a Expression Is a Seasonal and Estrus Timer in Reproductive Tissues. Endocrinology 2016, 157, 2469–2478. [Google Scholar] [CrossRef]
- Harris, C.J.; Scheibe, M.; Wongpalee, S.P.; Liu, W.; Cornett, E.M.; Vaughan, R.M.; Li, X.; Chen, W.; Xue, Y.; Zhong, Z.; et al. A DNA methylation reader complex that enhances gene transcription. Science 2018, 362, 1182–1186. [Google Scholar] [CrossRef]
- Halpern, K.B.; Vana, T.; Walker, M.D. Paradoxical Role of DNA Methylation in Activation of FoxA2 Gene Expression during Endoderm Development. J. Biol. Chem. 2014, 289, 23882–23892. [Google Scholar] [CrossRef]
- Wan, J.; Oliver, V.F.; Wang, G.; Zhu, H.; Zack, D.J.; Merbs, S.L.; Qian, J. Characterization of tissue-specific differential DNA methylation suggests distinct modes of positive and negative gene expression regulation. BMC Genom. 2015, 16, 49. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.F.; Xu, S.S.; Ren, X.; Lv, F.H.; Xie, X.L.; Zhao, Y.X.; Zhang, M.; Shen, Z.Q.; Ren, Y.L.; Gao, L.; et al. A genome-wide association study reveals candidate genes for the supernumerary nipple phenotype in sheep (Ovis aries). Anim. Genet. 2017, 48, 570–579. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; He, X.; Liu, W.; Yang, S.; Wang, L.; Li, W.; Wu, H.; Tang, S.; Ni, X.; Wang, J.; et al. Bi-allelic Mutations in TTC29 Cause Male Subfertility with Asthenoteratospermia in Humans and Mice. Am. J. Hum. Genet. 2019, 105, 1168–1181. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Names | Sequences (5′-3′) | Product Length (bp) | Gene Bank | |
|---|---|---|---|---|
| Forward | Reverse | |||
| GAPDH | CGGCACAGTCAAGGCAGAGAAC | CACGTACTCAGCACCAGCATCAC | 115 | XM_005680968.3 |
| DNMT1 | CCTGACTCCACCTACGAAGACC | CTACTTGCTCCACCACGAACTG | 127 | XM_018051019.1 |
| DNMT3A | CCGCATTGTGTCTTGGTGGATG | AGAACTTGCCGTCTCCGAACC | 86 | XM_018055548.1 |
| DNMT3B | TTGACTTGGTGATTGGTGGAAGC | CGAGTGTAATTCAGCAGGTGGTAG | 124 | XM_018057711.1 |
| PGF | TGAGACTGTTCACTTGCTTCCTG | GCTGCGGCTCCACACTTG | 131 | XM_005686088.3 |
| EPHA6 | AGGATACAGTGGCTACAGTCAGAAG | ACGGCTGCGGTGGCTATC | 105 | XM_013972057.2 |
| NFASC | ACGACAGCCTGGTGGACTATGG | TCCGTCTCCTCCTTGTCCTTCTTG | 103 | XM_018060022.1 |
| NDRG4 | CCAACCACCACGACCTTCCTG | AGCCTTCGGTCCTTCAAGTATGC | 137 | XM_018062073.1 |
| LRP1B | GCACTGCGACTCTGATGATGACTG | ATCTGCCACTGGAACAACTGAACTG | 98 | XM_018062463.1 |
| LOC102181552 | TGTTCCTGCTGCTTCCAGATGG | GTACACCTCCACGTCATCCTCAC | 134 | XM_005697318.2 |
| CFAP43 | GCAAGGAAGCAGGAGGAGAGG | CCGCCAGTAAGTTGTCATAGTGTTC | 100 | XM_018041743.1 |
| TTC29 | GCATTAGCAGTCCTCAACACTTACG | CCGTGGCTTCATAGGCTCTCC | 85 | XM_005691202.2 |
| PDE11A | CTCTATGGAACCTCTGCCACCTTG | ATGTTGTGACCCTCGCTCTGAAG | 80 | XM_005675916.3 |
| Region | Sequences (5′-3′) | Product Length (bp) | Associate Gene | Gene Bank |
|---|---|---|---|---|
| chr18:27599410-27599622 | AAATATTTGTGTTTGGAGTTATTTT | 213 | NDRG4 | NC_030825.1 |
| TACCCCAATCTTTATTTAAATTCCC | ||||
| chr24:62258787_62258914 | TTTAGATTTAGTGATTAGATTAGTGGT | 128 | SERPINB2 | NC_030831.1 |
| AACAATAAAACCTAACTCCATACC | ||||
| chr2:79444158-79444322 | AAAATTTATGTAAATGTTAGAAGTTT | 165 | LRP1B | NC_030809.1 |
| AATAATTAAATAACATCACCAACTC | ||||
| chr1:39785850-39786019 | TTATTTAGGTTAGGGGTTGAGGG | 170 | EPHA6 | NC_030808.1 |
| ACACTAACAAAACCAAAAACAAAAC |
| Groups | Sample | Clean Reads 1 | Q30 1 (%) | Total Mapping Reads 1 (%) | Unique Mapping Ratio 1 (%) | Bisulfite Conversion Rate 1 (%) | Total_mC 1 (%) |
|---|---|---|---|---|---|---|---|
| HP | HP-O1 | 1139474436 | 91.39 | 85.54 | 79.17 | 99.54 | 3.50 |
| HP-O2 | 1148095570 | 91.62 | 83.5 | 77.57 | 99.51 | 3.43 | |
| HP-O3 | 1124419076 | 91.21 | 84.53 | 78.40 | 99.51 | 3.55 | |
| HP-O4 | 1185419974 | 91.31 | 80.05 | 74.17 | 99.47 | 3.30 | |
| LP | LP-O1 | 1234628940 | 91.94 | 83.81 | 77.90 | 99.53 | 3.24 |
| LP-O2 | 1120935898 | 90.94 | 83.48 | 77.42 | 99.53 | 3.48 | |
| LP-O3 | 1341873656 | 91.21 | 84.18 | 77.81 | 99.51 | 2.98 | |
| LP-O4 | 1042249584 | 90.16 | 82.49 | 76.80 | 99.47 | 3.66 |
| Group | Genes | Gene Counts |
|---|---|---|
| Hyper DMG-Hyper DEG | NFASC,GLT1D1,DOCK10,JAKMIP3,GRM5,GRIK2 | 6 |
| Hyper DMG-Hypo DEG | CR2, CDH20, CFAP43 [5,18,19], DAGLA, GRM8, ZNF804B, RELN, TRHDE, LOC102172692, CLEC9A | 10 |
| Hypo DMG-Hyper DEG | ISM1, MMP16, NCAM1, USH2A, GALNT9, VAT1L, UPK1A, LRP1B 1 [20,21], FMNL2, CTNND2, FSD2, MTCL1, SERPINB2 1 [22], ADAM12, DLG2, LOC102170378, GSTM3, GRIK2, TMEM176B | 19 |
| Hypo DMG-Hypo DEG | CADM2, EPHA6 1 [7], C1H3orf52, PGF 1 [23,24,25], ALDH1A2, LOC108637248, COL14A1, TTC29 1 [3], NDRG4 1 [26,27], HPN, ARHGAP27, PDE11A 1 [28,29,30], MFSD6, MYO7B, JAG2, CDH20, TRPM8, C3H1orf87, C3H1orf226, COL28A1, SUN3, CRACR2A, UNC5A, RFX2, FBXO16, LOC108634594 | 26 |
| Gene | Chr 1 | Start | End | Element | Meth-Direction | Meth Diff 1 | Q-Value |
|---|---|---|---|---|---|---|---|
| SERPINB2 | 24 | 62258801 | 62259000 | UP3K | Hypo | −0.331 | 1.09 × 10−3 |
| NDRG4 | 18 | 27599201 | 27599400 | UP3K | Hypo | −0.326 | 4.35 × 10−5 |
| CFAP43 | 26 | 27046601 | 27046800 | intro | Hyper | 0.435 | 1.65 × 10−2 |
| LRP1B | 2 | 79720201 | 79720400 | intro | Hypo | −0.312 | 2.81 × 10−2 |
| EPHA6 | 1 | 40088601 | 40088800 | intro | Hypo | −0.309 | 1.19 × 10−4 |
| TTC29 | 17 | 59385801 | 59386000 | intro | Hypo | −0.328 | 1.36 × 10−5 |
| PDE11A | 2 | 117418601 | 117418800 | intro | Hypo | −0.403 | 3.33 × 10−5 |
| PGF | 10 | 16913801 | 16914000 | exon | Hypo | −0.309 | 8.42 × 10−4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, C.; He, J.; Mao, J.; Ren, Y.; Liu, G.; Wei, C.; Zhang, G.; Tian, K.; Huang, X. Genome-Wide DNA Methylation Analysis and Functional Validation of Litter Size Traits in Jining Grey Goats. Genes 2024, 15, 353. https://doi.org/10.3390/genes15030353
Yang C, He J, Mao J, Ren Y, Liu G, Wei C, Zhang G, Tian K, Huang X. Genome-Wide DNA Methylation Analysis and Functional Validation of Litter Size Traits in Jining Grey Goats. Genes. 2024; 15(3):353. https://doi.org/10.3390/genes15030353
Chicago/Turabian StyleYang, Cunming, Junmin He, Jingyi Mao, Yifan Ren, Guifen Liu, Chen Wei, Guoping Zhang, Kechuan Tian, and Xixia Huang. 2024. "Genome-Wide DNA Methylation Analysis and Functional Validation of Litter Size Traits in Jining Grey Goats" Genes 15, no. 3: 353. https://doi.org/10.3390/genes15030353