
A Chromosome-Level Genome Assembly of the Non-Hematophagous Leech Whitmania pigra (Whitman 1884): Identification and Expression Analysis of Antithrombotic Genes

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. DNA and RNA Sequencing
2.2. Genome Assembling
2.3. Gene Prediction
2.4. Expression of Antithrombotic Genes
3. Results
3.1. Basic Information of Genome Assembly
3.2. Antithrombotic Genes and Proteins
3.3. Gene Expression
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Favaloro, E.J.; Gosselin, R.C.; Pasalic, L.; Lippi, G. Hemostasis and thrombosis: An overview focusing on associated laboratory testing to diagnose and help manage related disorders. Methods Mol. Biol. 2023, 2663, 3–38. [Google Scholar]
- WHO. The Top 10 Causes of Death. 2020. Available online: https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death (accessed on 9 December 2020).
- Li, T.; Yuan, D.; Yuan, J. Antithrombotic drugs-pharmacology and perspectives. Adv. Exp. Med. Biol. 2020, 1177, 101–131. [Google Scholar]
- Elantably, D.; Mourad, A.; Elantably, A.; Effat, M. Warfarin induced leukocytoclastic vasculitis: An extraordinary side effect. J. Thromb. Thrombolysis 2020, 49, 149–152. [Google Scholar] [CrossRef] [PubMed]
- Al-Husein, B.A.; Al-Azzam, S.I.; Alzoubi, K.H.; Khabour, O.F.; Nusair, M.B.; Alzayadeen, S. Investigating the effect of demographics, clinical characteristics, and polymorphism of MDR-1, CYP1A2, CYP3A4, and CYP3A5 on clopidogrel resistance. J. Cardiovasc. Pharmacol. 2018, 72, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Giahchi, F.; Mohammadi, M. Reteplase versus streptokinase in management of ST-segment elevation myocardial infarction; a letter to the editor. Adv. J. Emerg. Med. 2019, 3, e34. [Google Scholar]
- Sawyer, R.T. Leech Biology and Behaviour; Oxford University Press: Oxford, UK, 1986. [Google Scholar]
- Sket, B.; Trontelj, P. Global diversity of leeches (Hirudinea) in freshwater. Hydrobiologia 2008, 595, 129–137. [Google Scholar] [CrossRef]
- Li, F.G.; Shi, X.Y.; Yang, L.; Lu, X.; Qi, Y.; Li, P.; Yang, H.; Gao, W. Quantitative proteomics based bioactive proteins discovery and quality control of medicinal leeches. J. Ethnopharmac. 2023, 319, 117117. [Google Scholar] [CrossRef]
- Ma, C.J.; Li, X.; Chen, H. Research progress in the use of leeches for medical purposes. Tradit. Med. Res. 2021, 6, 56–69. [Google Scholar] [CrossRef]
- Borda, E.; Siddall, M.E. Arhynchobdellida (Annelida: Oligochaeta: Hirudinida): Phylogenetic relationships and evolution. Mol. Phylogenet. Evol. 2004, 30, 213–225. [Google Scholar] [CrossRef]
- Siddall, M.E.; Burreson, E.M. Phylogeny of the Euhirudinea: Independent evolution of blood feeding by leeches? Can. J. Zool. 1995, 73, 1048–1064. [Google Scholar] [CrossRef]
- Phillips, A.J.; Siddall, M.E. Poly-paraphyly of Hirudinidae: Many lineages of medicinal leeches. BMC Evol. Biol. 2009, 9, 246. [Google Scholar] [CrossRef]
- Yang, T. Fauna Sinica (Annelida: Hirudinea); Science Press: Beijing, China, 1996. [Google Scholar]
- Chinese Pharmacopoeia Commission. Pharmacopoeia of the People’s Republic of China; Medicine Science and Technology Press: Beijing, China, 2020. [Google Scholar]
- He, C.H.; Chen, X.Y.; Zhang, X.M.; Wang, L.; Hu, S.M. A textual research on Whitmania pigra Whitman as the origin of Hirudo. China Med. Her. 2021, 18, 112–115. [Google Scholar]
- Song, J.; Zhou, Q. Discussion on the sources of Shuizhi (leeches) in Chinese Pharmacopoeia. Shizhen J. Tradit. Chin. Med. Res. 1997, 8, 104. [Google Scholar]
- Ding, Y.Z.; Duan, T.X.; Shan, Y.; Wang, X.F.; Yang, H.; Yuan, R.J. Comparative studies on anti-thrombin activity and anticoagulant mechanism between Whitmania pigra Whitman and Hirudinaria manillensis. China Pharm. 2016, 19, 1621–1624. [Google Scholar]
- Li, W.; Liao, F.; Yin, X.; Peng, J.; Ou, X.; Zhang, Q.; Ding, J.; Yang, T. Experimental study on anti-platelet aggregation and anti-coagulation of seven spices of leech. Pharmacol. Clin. Chin. Mat. Med. 1997, 13, 32–34. [Google Scholar]
- Wang, X.; Gan, Q.C.; Hu, H.; Hao, J.; Wu, G.; Gao, Q.; Li, X.; Niu, M.; Wang, J.; Ma, L.; et al. Measurement of antiplatelet aggregation and potency of Hirudo. Acta Pharm. Sin. 2019, 54, 2178–2183. [Google Scholar]
- Müller, C.; Wang, Z.J.; Hamann, M.; Sponholz, D.; Hildebrandt, J.P. Life without blood: Molecular and functional analysis of hirudins and hirudin-like factors of the Asian non-hematophagous leech Whitmania pigra. J. Thromb. Haemost. 2022, 20, 1808–1817. [Google Scholar] [CrossRef]
- Simakov, O.; Marletaz, F.; Cho, S.J.; Edsinger-Gonzales, E.; Havlak, P.; Hellsten, U.; Kuo, D.H.; Larsson, T.; Lv, J.; Arendt, D.; et al. Insights into bilaterian evolution from three spiralian genomes. Nature 2013, 493, 526–531. [Google Scholar] [CrossRef]
- Babenko, V.V.; Podgorny, O.V.; Manuvera, V.A.; Kasianov, A.S.; Manolov, A.I.; Grafskaia, E.N.; Shirokov, D.A.; Kurdyumov, A.S.; Vinogradov, D.V.; Nikitina, A.S.; et al. Draft genome sequences of Hirudo medicinalis and salivary transcriptome of three closely related medicinal leeches. BMC Genom. 2020, 21, 331. [Google Scholar]
- Kvist, S.; Manzano-Marín, A.; de Carle, D.; Trontelj, P.; Siddall, M.E. Draft genome of the European medicinal leech Hirudo medicinalis (Annelida, Clitellata, Hirudiniformes) with emphasis on anticoagulants. Sci. Rep. 2020, 10, 9885. [Google Scholar] [CrossRef] [PubMed]
- Guan, D.L.; Yang, J.; Liu, Y.K.; Li, Y.; Mi, D.; Ma, L.B.; Wang, Z.Z.; Xu, S.Q.; Qiu, Q. Draft genome of the Asian buffalo leech Hirudinaria manillensis. Front. Genet. 2020, 10, 1321. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.H.; Wang, X.B.; Feng, T.; Rehman, S.U.; Yan, X.Y.; Shan, H.Q.; Ma, X.C.; Zhou, W.G.; Xu, W.H.; Lu, L.Y.; et al. Molecular mechanisms underlying hematophagia revealed by comparative analyses of leech genomes. Gigascience 2023, 12, giad023. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.C.; Zhao, F.; Huang, Z.H.; Hu, Q.M.; Meng, R.Y.; Lin, Y.Q.; Qi, J.X.; Lin, G.H. Revisiting the Asian buffalo leech (Hirudinaria manillensis) genome: Focus on antithrombotic genes and their corresponding proteins. Genes 2023, 14, 2068. [Google Scholar] [CrossRef]
- Tong, L.; Dai, S.X.; Kong, D.J.; Yang, P.P.; Tong, X.; Tong, X.R.; Bi, X.X.; Su, Y.; Zhao, Y.Q.; Liu, Z.C. The genome of medicinal leech (Whitmania pigra) and comparative genomic study for exploration of bioactive ingredients. BMC Genom. 2022, 23, 76. [Google Scholar] [CrossRef]
- Hu, J.; Wang, Z.; Sun, Z.; Hu, B.; Ayoola, A.O.; Liang, F.; Li, J.; Sandoval, J.R.; Cooper, D.N.; Ye, K.; et al. An efficient error correction and accurate assembly tool for noisy long reads. bioRxiv 2023. [Google Scholar] [CrossRef]
- Hu, J.; Fan, J.; Sun, Z.; Liu, S. NextPolish: A fast and efficient genome polishing tool for long-read assembly. Bioinformatics 2020, 36, 2253–2255. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.X.; McCarthy, S.A.; Durbin, R. YaHS: Yet another Hi-C scaffolding tool. Bioinformatics 2023, 39, btac808. [Google Scholar] [CrossRef]
- Durand, N.C.; Robinson, J.T.; Shamim, M.S.; Machol, I.; Mesirov, J.P.; Lander, E.S.; Aiden, E.L. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst. 2016, 3, 99–101. [Google Scholar] [CrossRef]
- Durand, N.C.; Shamim, M.S.; Machol, I.; Rao, S.S.P.; Huntley, M.H.; Lander, E.S.; Aiden, E.L. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 2016, 3, 95–98. [Google Scholar] [CrossRef]
- Jin, J.J.; Yu, W.B.; Yang, J.B.; Song, Y.; dePamphilis, C.W.; Yi, T.S.; Li, D.Z. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef]
- Seppey, M.; Manni, M.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness. Methods Mol. Biol. 2019, 1962, 227–245. [Google Scholar]
- Rhie, A.; Walenz, B.P.; Koren, S.; Phillippy, A.M. Merqury: Reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 2020, 21, 245. [Google Scholar] [CrossRef]
- Flynn, J.M.; Hubley, R.; Goubert, C.; Rosen, J.; Clark, A.G.; Feschotte, C.; Smit, A.F. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 2020, 117, 9451–9457. [Google Scholar] [CrossRef]
- Bao, W.D.; Kojima, K.K.; Kohany, O. Repbase update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 2015, 6, 11. [Google Scholar] [CrossRef]
- Hoff, K.J.; Lange, S.; Lomsadze, A.; Borodovsky, M.; Stanke, M. BRAKER1: Unsupervised RNA-seq-based genome annotation with GeneMark-ET and AUGUSTUS. Bioinformatics 2016, 32, 767–769. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.D.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Tang, S.; Lomsadze, A.; Borodovsky, M. Identification of protein coding regions in RNA transcripts. Nucleic Acids Res. 2015, 43, e78. [Google Scholar] [CrossRef]
- Slater, G.S.C.; Birney, E. Automated generation of heuristics for biological sequence comparison. BMC Bioinform. 2005, 6, 31. [Google Scholar] [CrossRef]
- Dainat, J. AGAT: Another gff analysis toolkit to handle annotations in any GTF/GFF format (v0.7.0). Zenodo 2021. [Google Scholar] [CrossRef]
- Pertea, G.; Pertea, M. GFF utilities: GffRead and GffCompare. F1000Research 2020, 9, 304. [Google Scholar] [CrossRef]
- Teufel, F.; Armenteros, J.J.A.; Johansen, A.R.; Gislason, M.H.; Pihl, S.I.; Tsirigos, K.D.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 6.0 predicts all five types of signal peptides using protein language models. Nat. Biotechnol. 2022, 40, 1023–1025. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European molecular biology open software suite. Trends. Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef]
- Dunwiddie, C.; Thornberry, N.A.; Bull, H.G.; Sardana, M.; Friedman, P.A.; Jacobs, J.W.; Simpson, E. Antistasin, a leech-derived inhibitor of factor Xa. Kinetic analysis of enzyme inhibition and identification of the reactive site. J. Biol. Chem. 1989, 264, 16694–16699. [Google Scholar] [CrossRef] [PubMed]
- Marin, E.; Kornilov, D.A.; Bukhdruker, S.S.; Aleksenko, V.A.; Manuvera, V.A.; Zinovev, E.V.; Kovalev, K.V.; Shevtsov, M.B.; Talyzina, A.A.; Bobrovsky, P.A.; et al. Structural insights into thrombolytic activity of destabilase from medicinal leech. Sci. Rep. 2023, 13, 6641. [Google Scholar] [CrossRef]
- Castellano, I.; Merlino, A. γ-Glutamyltranspeptidases: Sequence, structure, biochemical properties, and biotechnological applications. Cell. Mol. Life Sci. 2012, 69, 3381–3394. [Google Scholar] [CrossRef]
- Zhao, F.; Jiang, K.; Su, T.J.; He, B.; Wu, Q.; Lin, G.H.; Huang, Z.H. Phylogenomics of common species in Hirudiniformes. J. Jinggangshan Univ. (Nat. Sci.) 2021, 42, 41–47. [Google Scholar]
- Qiao, N.; Bai, Y.; Wang, G.; Xu, S. Comparative morphology study on the jaws of three leech species of the genus Whitmania Blanchard. Sichuan J. Zool. 2013, 32, 526–529. [Google Scholar]
- Guan, S.X.; Yuan, Z.W.; Zhou, Y.B.; Zhang, Y.; Ye, X.L.; Hu, B. Comparative studies on anti-thrombus and anti-coagulation effects of Hirudo of different species. Chin. J. Hosp. Pharm. 2012, 32, 1093–1096. [Google Scholar]
- Betz, S.F. Disulfide bonds and the stability of globular proteins. Protein Sci. 1993, 2, 1551–1558. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
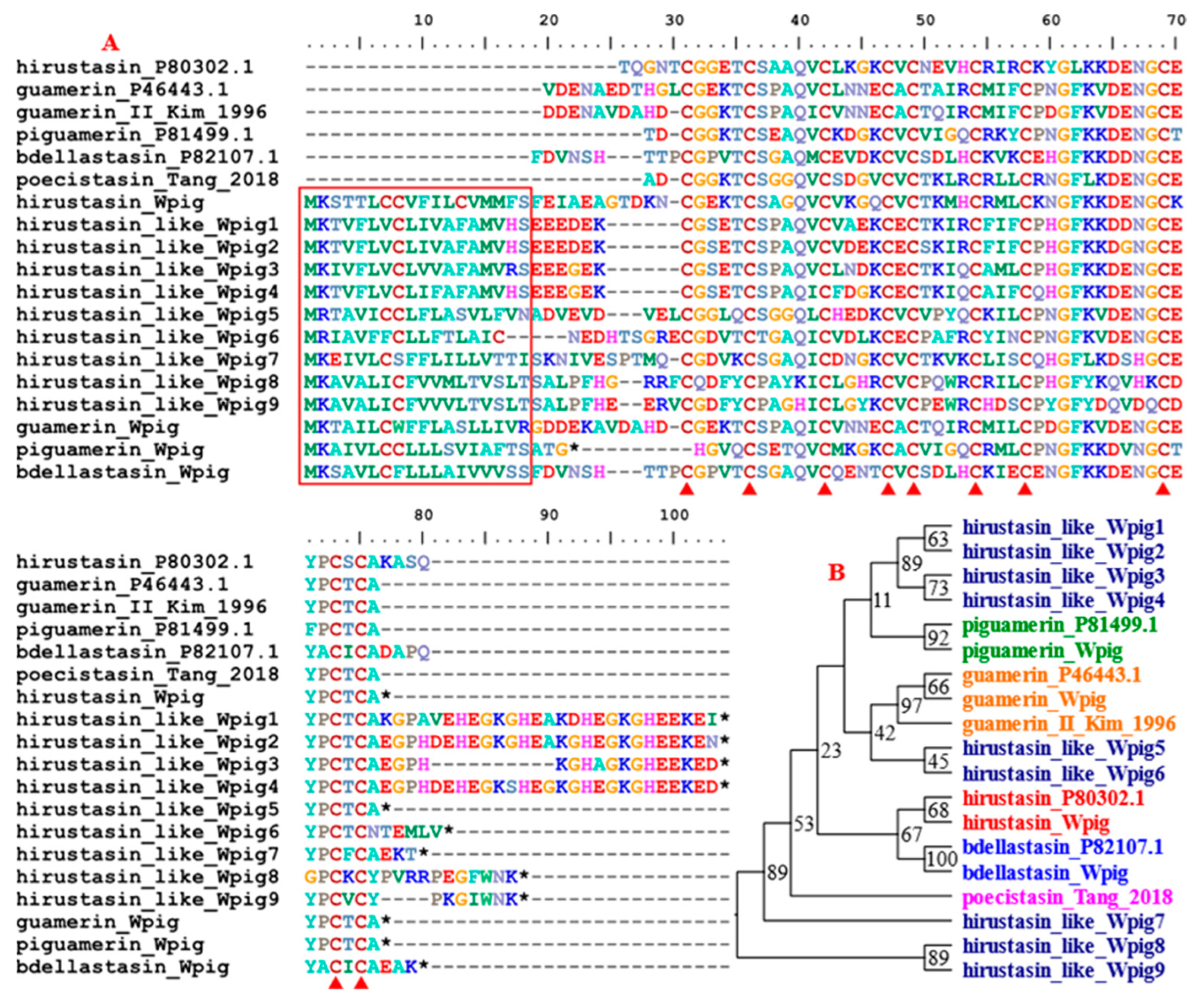{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Family | W. pigra | H. manillensis | Protein Function |
|---|---|---|---|
| hirudin | 7 | 5 | coagulation inhibitor |
| progranulin | 1 | 1 | coagulation inhibitor |
| antistasin | 2 | 2 | coagulation inhibitor |
| lefaxin | 3 | 3 | coagulation inhibitor |
| therostasin | 1 | 1 | coagulation inhibitor |
| hirustasin/hirustasin-like | 1/9 | 1/12 | coagulation inhibitor |
| guamerin | 1 | 1 | coagulation inhibitor |
| piguamerin | 1 | 1 | coagulation inhibitor |
| bdellastasin | 1 | 1 | coagulation inhibitor |
| poecistasin | 0 | 2 | coagulation inhibitor |
| eglin | 2 | 4 | coagulation inhibitor |
| bdellin | 1 | 1 | coagulation inhibitor |
| LDTI | 1 | 1 | coagulation inhibitor |
| HMEI | 20 | 18 | coagulation inhibitor |
| saratin | 11 | 2 | platelet aggregation inhibitor |
| apyrase | 3 | 5 | platelet aggregation inhibitor |
| lumbrokinase | 4 | 3 | platelet aggregation inhibitor |
| destabilase | 4 | 3 | fibrinolysis enhancer |
| GGT | 2 | 1 | fibrinolysis enhancer |
| LCI | 1 | 1 | fibrinolysis enhancer |
| hyaluronidase | 3 | 3 | tissue penetration enhancer |
| total | 79 | 72 | — |
| Query Protein | Target Species | Target Protein | Target Accession | Identity | Function |
|---|---|---|---|---|---|
| hirudin_Wpig1~4 | W. pigra | Wpig_V6 | USH09350.1 | 100% | inactive |
| hirudin_Wpig5 | W. pigra | Wpig_V2 | USH09353.1 | 98.72% | inactive |
| hirudin_Wpig6 | W. pigra | Wpig_V3 | USH09356.1 | 100% | inactive |
| hirudin_Wpig7 | W. pigra | Wpig_V1 | USH09346.1 | 100% | active |
| Species | Sample | Antithrombotic Genes | Non-Antithrombotic Genes |
|---|---|---|---|
| H. manillensis | Hman_SRR26151944 | 605.8 | 37.8 |
| Hman_SRR26541753 | 794.1 | 37.3 | |
| Hman_SRR26541752 | 675.9 | 37.6 | |
| Hman_SRR26541746 | 789.4 | 37.3 | |
| Hman_SRR15881208 | 742.3 | 37.5 | |
| Hman_SRR15881209 | 415.1 | 38.4 | |
| Hman_SRR15881210 | 739.5 | 37.5 | |
| Mean ± SD | 680.3 ± 134.2 | 37.6 ± 0.4 | |
| W. pigra | Wpig_SRR26513850 | 2199.1 | 34.3 |
| Wpig_SRR26541745 | 882.7 | 38.6 | |
| Wpig_SRR26541744 | 588.2 | 39.6 | |
| Wpig_SRR26541743 | 838.1 | 38.8 | |
| Wpig_SRR15881156 | 863.8 | 38.7 | |
| Wpig_SRR15881157 | 1514.5 | 36.6 | |
| Wpig_SRR15881159 | 811.9 | 38.9 | |
| Mean ± SD | 1099.8 ± 562.2 | 37.9 ± 1.8 |
| Gene Family | tTPM (Mean ± SD) | Mann-Whitney U test | ||
|---|---|---|---|---|
| H. manillensis | W. pigra | Z Value | p Value | |
| hirudin | 523.4 ± 577.8 | 12,942.4 ± 12,976.2 | −2.747 | 0.004 |
| progranulin | 230.8 ± 119.1 | 172.9 ± 187.6 | −1.214 | 0.259 |
| antistasin | 454.0 ± 419.5 | 8268.1 ± 7249.5 | −2.364 | 0.017 |
| lefaxin | 18,963.3 ± 5334 | 20,150.9 ± 12,370.5 | −0.064 | 1.000 |
| therostasin | 32.3 ± 53.1 | 34.1 ± 46.3 | −0.332 | 0.805 |
| Hirustasin # | 6398.5 ± 2887.8 | 6135.1 ± 4635.6 | −0.064 | 1.000 |
| eglin | 7050.9 ± 5701.4 | 197.2 ± 133.1 | −2.619 | 0.007 |
| bdellin | 2611.7 ± 1291.6 | 746.1 ± 707.6 | −2.747 | 0.004 |
| LDTI | 2312.8 ± 1960.7 | 27.9 ± 43.5 | −2.364 | 0.017 |
| HMEI | 3490.8 ± 2578.1 | 3618.2 ± 2670.2 | −0.064 | 1.000 |
| saratin | 1479.3 ± 1886.7 | 16,195.5 ± 23,270.1 | −2.619 | 0.007 |
| apyrase | 492.1 ± 597.7 | 25.2 ± 30.0 | −2.747 | 0.004 |
| lumbrokinase | 0.3 ± 0.3 | 7.2 ± 9.5 | −2.619 | 0.007 |
| destabilase | 3341.9 ± 3137.4 | 16,864.1 ± 14,048.7 | −2.875 | 0.002 |
| GGT | 17.5 ± 11.9 | 77.5 ± 86.3 | −1.214 | 0.259 |
| LCI | 1415.8 ± 1037.7 | 1333.9 ± 1602.4 | −0.192 | 0.902 |
| hyaluronidase | 166.6 ± 198.0 | 84.1 ± 34.9 | −0.447 | 0.710 |
| Species | Gene | TPM (Mean ± SD) | Anticoagulation |
|---|---|---|---|
| H. manillensis | hirudin_Hman1 | 176.0 ± 300.2 | active |
| hirudin_Hman2 | 238.8 ± 630.8 | active | |
| hirudin_Hman3 | 3.6 ± 5.6 | inactive | |
| hirudin_Hman4 | 105.0 ± 185.1 | inactive | |
| hirudin_Hman5 | 0.0 ± 0.0 | active | |
| W. pigra # | hirudin_Wpig1 | 1985.9 ± 2303.0 | inactive |
| hirudin_Wpig2 | 1985.9 ± 2303.0 | inactive | |
| hirudin_Wpig3 | 1985.9 ± 2303.0 | inactive | |
| hirudin_Wpig4 | 1985.9 ± 2303.0 | inactive | |
| hirudin_Wpig5 | 1892.2 ± 2371.9 | inactive | |
| hirudin_Wpig6 | 3102.4 ± 3593.7 | inactive | |
| hirudin_Wpig7 | 4.0 ± 8.0 | active |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Z.; Zhao, F.; Huang, Z.; He, B.; Liu, K.; Shi, F.; Zhao, Z.; Lin, G. A Chromosome-Level Genome Assembly of the Non-Hematophagous Leech Whitmania pigra (Whitman 1884): Identification and Expression Analysis of Antithrombotic Genes. Genes 2024, 15, 164. https://doi.org/10.3390/genes15020164
Liu Z, Zhao F, Huang Z, He B, Liu K, Shi F, Zhao Z, Lin G. A Chromosome-Level Genome Assembly of the Non-Hematophagous Leech Whitmania pigra (Whitman 1884): Identification and Expression Analysis of Antithrombotic Genes. Genes. 2024; 15(2):164. https://doi.org/10.3390/genes15020164
Chicago/Turabian StyleLiu, Zichao, Fang Zhao, Zuhao Huang, Bo He, Kaiqing Liu, Feng Shi, Zheng Zhao, and Gonghua Lin. 2024. "A Chromosome-Level Genome Assembly of the Non-Hematophagous Leech Whitmania pigra (Whitman 1884): Identification and Expression Analysis of Antithrombotic Genes" Genes 15, no. 2: 164. https://doi.org/10.3390/genes15020164