
SLCO1B1 Genetic Variation Influence on Atorvastatin Systemic Exposure in Pediatric Hypercholesterolemia
 , , ,
, , ,
Abstract
:1. Introduction
2. Methods
2.1. Subjects
2.2. Genetic Analysis
2.3. Study Design
2.4. Analytical Methods
2.5. Pharmacokinetic Parameters
2.6. Statistical Analyses
3. Results
3.1. Participant Demographics and Adverse Events
3.2. Drug Disposition Profiles
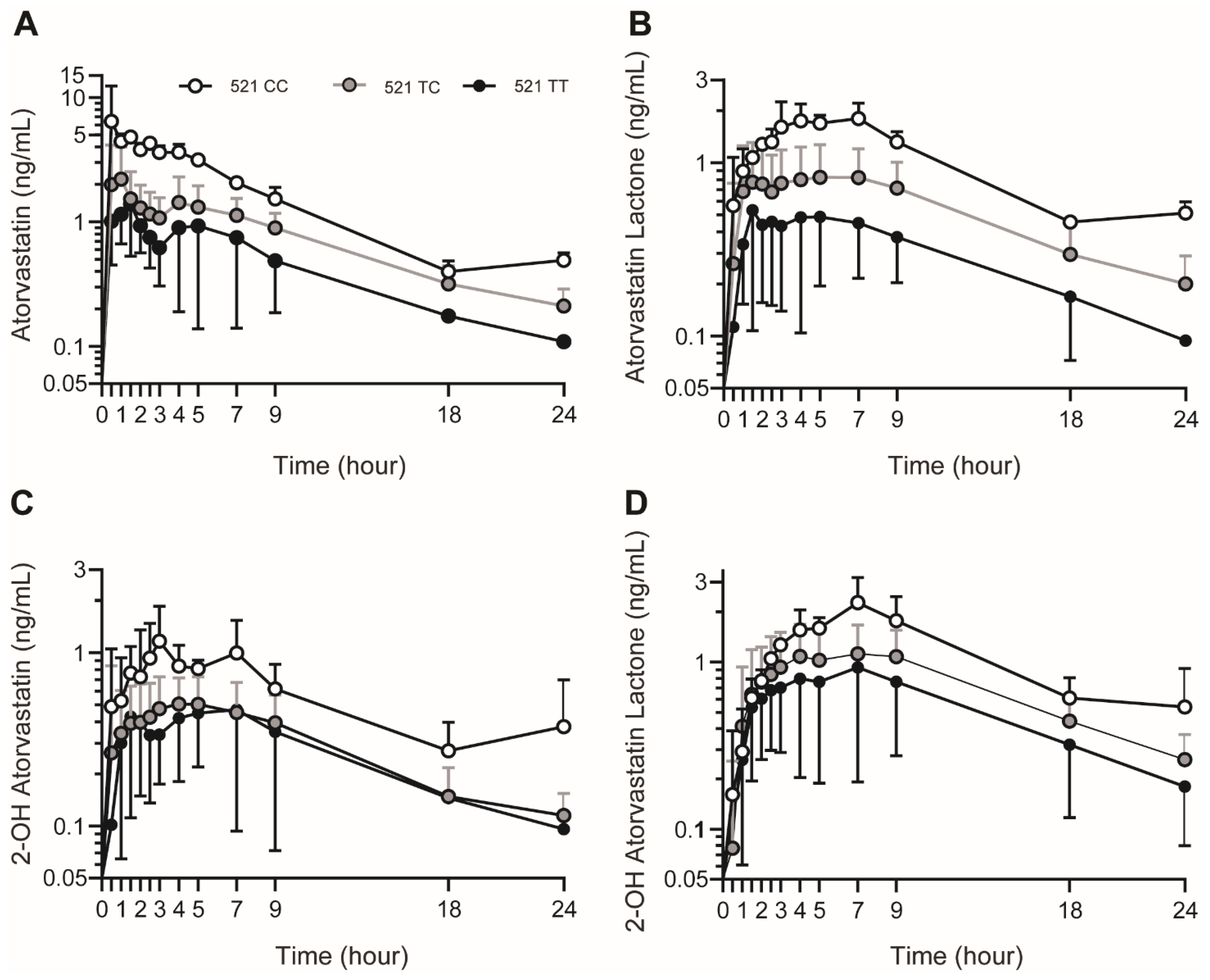
3.3. SLCO1B1 c.521T>C Genotype Influence on Plasma AVA and AVA Metabolite Concentrations
3.4. Demographic and Anthropometric Influence on Plasma AVA and AVA Metabolite Concentrations
3.5. Non-SLCO1B1 Drug Transporter Influence on Plasma AVA and AVA Metabolite Concentrations
3.6. Effect of CYP3A and UGT on Plasma AVA and AVA Metabolite Concentrations
3.7. Plasma AVA Concentration Multivariate Model
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- LipitorTM (Atorvastatin Calcium) [Package Insert]; Pfizer: New York, NY, USA, 1996; Revised 2020 September.
- Shitara, Y.; Sugiyama, Y. Pharmacokinetic and pharmacodynamic alterations of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors: Drug-drug interactions and interindividual differences in transporter and metabolic enzyme functions. Pharmacol. Ther. 2006, 112, 71–105. [Google Scholar] [CrossRef] [PubMed]
- Cilla, D.D., Jr.; Whitfield, L.R.; Gibson, D.M.; Sedman, A.J.; Posvar, E.L. Multiple-dose pharmacokinetics, pharmacodynamics, and safety of atorvastatin, an inhibitor of HMG-CoA reductase, in healthy subjects. Clin. Pharmacol. Ther. 1996, 60, 687–695. [Google Scholar] [CrossRef]
- Vildhede, A.; Karlgren, M.; Svedberg, E.K.; Wisniewski, J.R.; Lai, Y.; Noren, A.; Artursson, P. Hepatic uptake of atorvastatin: Influence of variability in transporter expression on uptake clearance and drug-drug interactions. Drug Metab. Dispos. 2014, 42, 1210–1218. [Google Scholar] [CrossRef] [PubMed]
- Lea, A.P.; McTavish, D. Atorvastatin. A review of its pharmacology and therapeutic potential in the management of hyperlipidaemias. Drugs 1997, 53, 828–847. [Google Scholar] [CrossRef]
- Lennernas, H. Clinical pharmacokinetics of atorvastatin. Clin. Pharmacokinet. 2003, 42, 1141–1160. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, W.; Kuhn, B.; Soldner, A.; Kirchner, G.; Sewing, K.F.; Kollman, P.A.; Benet, L.Z.; Christians, U. Lactonization is the critical first step in the disposition of the 3-hydroxy-3-methylglutaryl-CoA reductase inhibitor atorvastatin. Drug Metab. Dispos. 2000, 28, 1369–1378. [Google Scholar] [PubMed]
- Goosen, T.C.; Bauman, J.N.; Davis, J.A.; Yu, C.; Hurst, S.I.; Williams, J.A.; Loi, C.M. Atorvastatin glucuronidation is minimally and nonselectively inhibited by the fibrates gemfibrozil, fenofibrate, and fenofibric acid. Drug Metab. Dispos. 2007, 35, 1315–1324. [Google Scholar] [CrossRef]
- Wu, X.; Whitfield, L.R.; Stewart, B.H. Atorvastatin transport in the Caco-2 cell model: Contributions of P-glycoprotein and the proton-monocarboxylic acid co-transporter. Pharm. Res. 2000, 17, 209–215. [Google Scholar] [CrossRef]
- Hirano, M.; Maeda, K.; Matsushima, S.; Nozaki, Y.; Kusuhara, H.; Sugiyama, Y. Involvement of BCRP (ABCG2) in the biliary excretion of pitavastatin. Mol. Pharmacol. 2005, 68, 800–807. [Google Scholar] [CrossRef]
- Botti, R.E.; Triscari, J.; Pan, H.Y.; Zayat, J. Concentrations of pravastatin and lovastatin in cerebrospinal fluid in healthy subjects. Clin. Neuropharmacol. 1991, 14, 256–261. [Google Scholar] [CrossRef]
- Johnson-Anuna, L.N.; Eckert, G.P.; Keller, J.H.; Igbavboa, U.; Franke, C.; Fechner, T.; Schubert-Zsilavecz, M.; Karas, M.; Muller, W.E.; Wood, W.G. Chronic administration of statins alters multiple gene expression patterns in mouse cerebral cortex. J. Pharmacol. Exp. Ther. 2005, 312, 786–793. [Google Scholar] [CrossRef] [PubMed]
- Pierno, S.; De Luca, A.; Tricarico, D.; Roselli, A.; Natuzzi, F.; Ferrannini, E.; Laico, M.; Camerino, D.C. Potential risk of myopathy by HMG-CoA reductase inhibitors: A comparison of pravastatin and simvastatin effects on membrane electrical properties of rat skeletal muscle fibers. J. Pharmacol. Exp. Ther. 1995, 275, 1490–1496. [Google Scholar]
- Bruckert, E.; Hayem, G.; Dejager, S.; Yau, C.; Begaud, B. Mild to moderate muscular symptoms with high-dosage statin therapy in hyperlipidemic patients—The PRIMO study. Cardiovasc. Drugs Ther. 2005, 19, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Masters, B.A.; Palmoski, M.J.; Flint, O.P.; Gregg, R.E.; Wang-Iverson, D.; Durham, S.K. In vitro myotoxicity of the 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors, pravastatin, lovastatin, and simvastatin, using neonatal rat skeletal myocytes. Toxicol. Appl. Pharmacol. 1995, 131, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, K.; Kuriyama, M.; Sonoda, Y.; Yoshidome, H.; Nakagawa, H.; Fujiyama, J.; Higuchi, I.; Osame, M. Myopathy induced by HMG-CoA reductase inhibitors in rabbits: A pathological, electrophysiological, and biochemical study. Toxicol. Appl. Pharmacol. 1998, 152, 99–106. [Google Scholar] [CrossRef]
- Gadbut, A.P.; Caruso, A.P.; Galper, J.B. Differential sensitivity of C2-C12 striated muscle cells to lovastatin and pravastatin. J. Mol. Cell. Cardiol. 1995, 27, 2397–2402. [Google Scholar] [CrossRef]
- McCrindle, B.W.; Ose, L.; Marais, A.D. Efficacy and safety of atorvastatin in children and adolescents with familial hypercholesterolemia or severe hyperlipidemia: A multicenter, randomized, placebo-controlled trial. J. Pediatr. 2003, 143, 74–80. [Google Scholar] [CrossRef]
- Gandelman, K.; Glue, P.; Laskey, R.; Jones, J.; LaBadie, R.; Ose, L. An eight-week trial investigating the efficacy and tolerability of atorvastatin for children and adolescents with heterozygous familial hypercholesterolemia. Pediatr. Cardiol. 2011, 32, 433–441. [Google Scholar] [CrossRef]
- Argent, E.; Kainer, G.; Aitken, M.; Rosenberg, A.R.; Mackie, F.E. Atorvastatin treatment for hyperlipidemia in pediatric renal transplant recipients. Pediatr. Transplant. 2003, 7, 38–42. [Google Scholar] [CrossRef]
- Kameyama, Y.; Yamashita, K.; Kobayashi, K.; Hosokawa, M.; Chiba, K. Functional characterization of SLCO1B1 (OATP-C) variants, SLCO1B1*5, SLCO1B1*15 and SLCO1B1*15+C1007G, by using transient expression systems of HeLa and HEK293 cells. Pharmacogenet. Genom. 2005, 15, 513–522. [Google Scholar] [CrossRef]
- Hsiang, B.; Zhu, Y.; Wang, Z.; Wu, Y.; Sasseville, V.; Yang, W.P.; Kirchgessner, T.G. A novel human hepatic organic anion transporting polypeptide (OATP2). Identification of a liver-specific human organic anion transporting polypeptide and identification of rat and human hydroxymethylglutaryl-CoA reductase inhibitor transporters. J. Biol. Chem. 1999, 274, 37161–37168. [Google Scholar] [CrossRef]
- Pasanen, M.K.; Fredrikson, H.; Neuvonen, P.J.; Niemi, M. Different effects of SLCO1B1 polymorphism on the pharmacokinetics of atorvastatin and rosuvastatin. Clin. Pharmacol. Ther. 2007, 82, 726–733. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Lee, M.G.; Lim, L.A.; Jang, S.B.; Chung, J.Y. Effects of SLCO1B1 and ABCB1 genotypes on the pharmacokinetics of atorvastatin and 2-hydroxyatorvastatin in healthy Korean subjects. Int. J. Clin. Pharmacol. Ther. 2010, 48, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Birmingham, B.K.; Bujac, S.R.; Elsby, R.; Azumaya, C.T.; Wei, C.; Chen, Y.; Mosqueda-Garcia, R.; Ambrose, H.J. Impact of ABCG2 and SLCO1B1 polymorphisms on pharmacokinetics of rosuvastatin, atorvastatin and simvastatin acid in Caucasian and Asian subjects: A class effect? Eur. J. Clin. Pharmacol. 2015, 71, 341–355. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, L.B.; Gong, L.; Lee, S.B.; Wagner, J.B.; Zhou, X.; Sangkuhl, K.; Adams, S.M.; Straka, R.J.; Empey, P.E.; Boone, E.C.; et al. PharmVar GeneFocus: SLCO1B1. Clin. Pharmacol. Ther. 2023, 113, 782–793. [Google Scholar] [CrossRef] [PubMed]
- Gaedigk, A.; Casey, S.T.; Whirl-Carrillo, M.; Miller, N.A.; Klein, T.E. Pharmacogene Variation Consortium: A Global Resource and Repository for Pharmacogene Variation. Clin. Pharmacol. Ther. 2021, 110, 542–545. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.B.; Abdel-Rahman, S.; Van Haandel, L.; Gaedigk, A.; Gaedigk, R.; Raghuveer, G.; Kauffman, R.; Leeder, J.S. Impact of SLCO1B1 Genotype on Pediatric Simvastatin Acid Pharmacokinetics. J. Clin. Pharmacol. 2018, 58, 823–833. [Google Scholar] [CrossRef]
- Wagner, J.B.; Abdel-Rahman, S.; Gaedigk, R.; Gaedigk, A.; Raghuveer, G.; Staggs, V.S.; Kauffman, R.; Van Haandel, L.; Leeder, J.S. Impact of Genetic Variation on Pravastatin Systemic Exposure in Pediatric Hypercholesterolemia. Clin. Pharmacol. Ther. 2019, 105, 1501–1512. [Google Scholar] [CrossRef]
- Niemi, M.; Schaeffeler, E.; Lang, T.; Fromm, M.F.; Neuvonen, M.; Kyrklund, C.; Backman, J.T.; Kerb, R.; Schwab, M.; Neuvonen, P.J.; et al. High plasma pravastatin concentrations are associated with single nucleotide polymorphisms and haplotypes of organic anion transporting polypeptide-C (OATP-C, SLCO1B1). Pharmacogenetics 2004, 14, 429–440. [Google Scholar] [CrossRef]
- Wagner, J.B.; Abdel-Rahman, S.; Gaedigk, A.; Gaedigk, R.; Raghuveer, G.; Staggs, V.S.; Van Haandel, L.; Steven Leeder, J. Impact of SLCO1B1 genetic variation on rosuvastatin systemic exposure in pediatric hypercholesterolemia. Clin. Transl. Sci. 2020, 13, 628–637. [Google Scholar] [CrossRef]
- Keskitalo, J.E.; Kurkinen, K.J.; Neuvoneni, P.J.; Niemi, M. ABCB1 haplotypes differentially affect the pharmacokinetics of the acid and lactone forms of simvastatin and atorvastatin. Clin. Pharmacol. Ther. 2008, 84, 457–461. [Google Scholar] [CrossRef]
- Keskitalo, J.E.; Zolk, O.; Fromm, M.F.; Kurkinen, K.J.; Neuvonen, P.J.; Niemi, M. ABCG2 polymorphism markedly affects the pharmacokinetics of atorvastatin and rosuvastatin. Clin. Pharmacol. Ther. 2009, 86, 197–203. [Google Scholar] [CrossRef]
- Kivisto, K.T.; Niemi, M.; Schaeffeler, E.; Pitkala, K.; Tilvis, R.; Fromm, M.F.; Schwab, M.; Eichelbaum, M.; Strandberg, T. Lipid-lowering response to statins is affected by CYP3A5 polymorphism. Pharmacogenetics 2004, 14, 523–525. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Guo, Y.; Wrighton, S.A.; Cooke, G.E.; Sadee, W. Intronic polymorphism in CYP3A4 affects hepatic expression and response to statin drugs. Pharmacogenom. J. 2011, 11, 274–286. [Google Scholar] [CrossRef] [PubMed]
- Riedmaier, S.; Klein, K.; Hofmann, U.; Keskitalo, J.E.; Neuvonen, P.J.; Schwab, M.; Niemi, M.; Zanger, U.M. UDP-glucuronosyltransferase (UGT) polymorphisms affect atorvastatin lactonization in vitro and in vivo. Clin. Pharmacol. Ther. 2010, 87, 65–73. [Google Scholar] [CrossRef]
- Twist, G.P.; Gaedigk, A.; Miller, N.A.; Farrow, E.G.; Willig, L.K.; Dinwiddie, D.L.; Petrikin, J.E.; Soden, S.E.; Herd, S.; Gibson, M.; et al. Constellation: A tool for rapid, automated phenotype assignment of a highly polymorphic pharmacogene, CYP2D6, from whole-genome sequences. NPJ Genom. Med. 2016, 1, 15007. [Google Scholar] [CrossRef] [PubMed]
- Pasanen, M.K.; Neuvonen, M.; Neuvonen, P.J.; Niemi, M. SLCO1B1 polymorphism markedly affects the pharmacokinetics of simvastatin acid. Pharmacogenet. Genom. 2006, 16, 873–879. [Google Scholar] [CrossRef]
- Schwarz, U.I.; Meyer zu Schwabedissen, H.E.; Tirona, R.G.; Suzuki, A.; Leake, B.F.; Mokrab, Y.; Mizuguchi, K.; Ho, R.H.; Kim, R.B. Identification of novel functional organic anion-transporting polypeptide 1B3 polymorphisms and assessment of substrate specificity. Pharmacogenet. Genom. 2011, 21, 103–114. [Google Scholar] [CrossRef]
- Niemi, M. Transporter pharmacogenetics and statin toxicity. Clin. Pharmacol. Ther. 2010, 87, 130–133. [Google Scholar] [CrossRef]
- Leslie, E.M.; Deeley, R.G.; Cole, S.P. Multidrug resistance proteins: Role of P-glycoprotein, MRP1, MRP2, and BCRP (ABCG2) in tissue defense. Toxicol. Appl. Pharmacol. 2005, 204, 216–237. [Google Scholar] [CrossRef]
- Anderson, E.L.; Howe, L.D.; Jones, H.E.; Higgins, J.P.; Lawlor, D.A.; Fraser, A. The Prevalence of Non-Alcoholic Fatty Liver Disease in Children and Adolescents: A Systematic Review and Meta-Analysis. PLoS ONE 2015, 10, e0140908. [Google Scholar] [CrossRef] [PubMed]
- Angulo, P. Nonalcoholic fatty liver disease. N. Engl. J. Med. 2002, 346, 1221–1231. [Google Scholar] [CrossRef] [PubMed]
- Fisher, C.D.; Lickteig, A.J.; Augustine, L.M.; Oude Elferink, R.P.; Besselsen, D.G.; Erickson, R.P.; Cherrington, N.J. Experimental non-alcoholic fatty liver disease results in decreased hepatic uptake transporter expression and function in rats. Eur. J. Pharmacol. 2009, 613, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Lake, A.D.; Novak, P.; Fisher, C.D.; Jackson, J.P.; Hardwick, R.N.; Billheimer, D.D.; Klimecki, W.T.; Cherrington, N.J. Analysis of global and absorption, distribution, metabolism, and elimination gene expression in the progressive stages of human nonalcoholic fatty liver disease. Drug Metab. Dispos. 2011, 39, 1954–1960. [Google Scholar] [CrossRef] [PubMed]
- Clarke, J.D.; Hardwick, R.N.; Lake, A.D.; Canet, M.J.; Cherrington, N.J. Experimental nonalcoholic steatohepatitis increases exposure to simvastatin hydroxy acid by decreasing hepatic organic anion transporting polypeptide expression. J. Pharmacol. Exp. Ther. 2014, 348, 452–458. [Google Scholar] [CrossRef]
- Clarke, J.D.; Hardwick, R.N.; Lake, A.D.; Lickteig, A.J.; Goedken, M.J.; Klaassen, C.D.; Cherrington, N.J. Synergistic interaction between genetics and disease on pravastatin disposition. J. Hepatol. 2014, 61, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Kulkarni, S.R.; Li, L.; Slitt, A.L. UDP-glucuronosyltransferase expression in mouse liver is increased in obesity- and fasting-induced steatosis. Drug Metab. Dispos. 2012, 40, 259–266. [Google Scholar] [CrossRef]
- Hardwick, R.N.; Ferreira, D.W.; More, V.R.; Lake, A.D.; Lu, Z.; Manautou, J.E.; Slitt, A.L.; Cherrington, N.J. Altered UDP-glucuronosyltransferase and sulfotransferase expression and function during progressive stages of human nonalcoholic fatty liver disease. Drug Metab. Dispos. 2013, 41, 554–561. [Google Scholar] [CrossRef]
- Kearney, A.S.; Crawford, L.F.; Mehta, S.C.; Radebaugh, G.W. The interconversion kinetics, equilibrium, and solubilities of the lactone and hydroxyacid forms of the HMG-CoA reductase inhibitor, CI-981. Pharm. Res. 1993, 10, 1461–1465. [Google Scholar] [CrossRef]
- Quitadamo, P.; Zenzeri, L.; Mozzillo, E.; Cuccurullo, I.; Rocco, A.; Franzese, A.; Nardone, G.; Staiano, A. Gastric Emptying Time, Esophageal pH-Impedance Parameters, Quality of Life, and Gastrointestinal Comorbidity in Obese Children and Adolescents. J. Pediatr. 2018, 194, 94–99. [Google Scholar] [CrossRef]
- Buchholz, V.; Berkenstadt, H.; Goitein, D.; Dickman, R.; Bernstine, H.; Rubin, M. Gastric emptying is not prolonged in obese patients. Surg. Obes. Relat. Dis. 2013, 9, 714–717. [Google Scholar] [CrossRef] [PubMed]
- Wright, R.A.; Krinsky, S.; Fleeman, C.; Trujillo, J.; Teague, E. Gastric emptying and obesity. Gastroenterology 1983, 84, 747–751. [Google Scholar] [CrossRef] [PubMed]
- Glasbrenner, B.; Pieramico, O.; Brecht-Krauss, D.; Baur, M.; Malfertheiner, P. Gastric emptying of solids and liquids in obesity. Clin. Investig. 1993, 71, 542–546. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SLCO1B1 c.521T/T (n = 13) | SLCO1B1 c.521T/C (n = 13) | SLCO1B1 c.521C/C (n = 2) | |
|---|---|---|---|
| Age (years) | 15.1 (±3.0) | 14.9 (±2.9) | 14.3 (±4.3) |
| Weight (kg) | 85.7 (±34.5) | 87.0 (±26.0) | 62.2 (±1.6) |
| Height (cm) | 164.1 (±8.5) | 165.3 (±11.8) | 157.6 (±19.8) |
| BMI (kg/m2) | 31.6 (±12.0) | 31.2 (±6.9) | 25.6 (±5.7) |
| Gender | |||
| Female | 6 | 6 | 1 |
| Male | 7 | 7 | 1 |
| Ethnicity | |||
| White, nonHispanic | 7 | 7 | 1 |
| White, Hispanic | 5 | 5 | 1 |
| African-American | 1 | 1 | 0 |
| Tanner Breast/Testicular | |||
| Stage 1 | 0 | 0 | 0 |
| Stage 2 | 2 | 2 | 1 |
| Stage 3 | 0 | 1 | 0 |
| Stage 4 | 4 | 0 | 0 |
| Stage 5 | 7 | 10 | 1 |
| Pubic | |||
| Stage 1 | 1 | 0 | 0 |
| Stage 2 | 0 | 3 | 1 |
| Stage 3 | 1 | 0 | 0 |
| Stage 4 | 3 | 0 | 0 |
| Stage 5 | 8 | 10 | 1 |
| Dose (mg/kg) | 0.14 (±0.06) | 0.13 (±0.04) | 0.16 (±0.00) |
| SLCO1B1 c.521T/T (n = 13) | SLCO1B1 c.521T/C (n = 13) | SLCO1B1 c.521C/C (n = 2) | p | |
|---|---|---|---|---|
| Atorvastatin | ||||
| Cmax (ng/mL) | 1.8 (±0.7) | 3.0 (±1.5) | 6.4 (±3.3) | 0.01 |
| tmax (h) | 1.0 (0.5–5.0) | 1.0 (0.5–7.0) | 1.0 (0.5–1.5) | N/A |
| AUC0–24 (ng·hr/mL) | 11.3 (±5.1) | 19.1 (±4.5) | 32.0 (±5.2) | 0.001 |
| AtorvastatinLactone | ||||
| Cmax (ng/mL) | 0.7 (±0.4) | 1.1 (±0.3) | 1.5 (±0.4) | 0.007 |
| tmax (h) | 2.5 (1.0–9.2) | 4.0 (1.0–9.0) | 6.0 (5.0–7.0) | N/A |
| AUC0–24 (ng·hr/mL) | 7.2 (±4.8) | 13.0 (±10.1) | 27.8 (±2.3) | 0.03 |
| 2-Hydroxy Atorvastatin | ||||
| Cmax (ng/mL) | 0.7 (±0.2) | 0.8 (±0.3) | 1.3 (±0.1) | 0.09 |
| tmax (h) | 2.0 (1.0–7.0) | 3.0 (0.5–7.0) | 5.0 (3.0–7.0) | N/A |
| AUC0–24 (ng·hr/mL) | 6.2 (±2.8) | 7.5 (±3.2) | 10.8 (±2.2) | 0.14 |
| 2-Hydroxy Atorvastatin Lactone | ||||
| Cmax (ng/mL) | 1.0 (±0.5) | 1.3 (±0.4) | 1.9 (±0.8) | 0.11 |
| tmax (h) | 7.0 (1.5–9.2) | 5.0 (2.0–7.0) | 7.0 (7.0–7.0) | N/A |
| AUC0–24 (ng·hr/mL) | 14.0 (±11.9) | 17.4 (±12.7) | 31.0 (±9.4) | 0.19 |
| Explanatory Variable | β (95% CI) | p |
|---|---|---|
| BMI z-score | 1.4 (−0.4, 3.1) | 0.114 |
| Male sex | 1.7 (−2.2, 5.6) | 0.378 |
| Tanner score | 0.5 (−1.1, 2.2) | 0.524 |
| SLCO1B1 c.521 number of “C” alleles | 9.0 (6.0, 12.0) | <0.001 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wagner, J.B.; Abdel-Rahman, S.; Raghuveer, G.; Gaedigk, A.; Boone, E.C.; Gaedigk, R.; Staggs, V.S.; Reed, G.A.; Zhang, N.; Leeder, J.S. SLCO1B1 Genetic Variation Influence on Atorvastatin Systemic Exposure in Pediatric Hypercholesterolemia. Genes 2024, 15, 99. https://doi.org/10.3390/genes15010099
Wagner JB, Abdel-Rahman S, Raghuveer G, Gaedigk A, Boone EC, Gaedigk R, Staggs VS, Reed GA, Zhang N, Leeder JS. SLCO1B1 Genetic Variation Influence on Atorvastatin Systemic Exposure in Pediatric Hypercholesterolemia. Genes. 2024; 15(1):99. https://doi.org/10.3390/genes15010099
Chicago/Turabian StyleWagner, Jonathan B., Susan Abdel-Rahman, Geetha Raghuveer, Andrea Gaedigk, Erin C. Boone, Roger Gaedigk, Vincent S. Staggs, Gregory A. Reed, Na Zhang, and J. Steven Leeder. 2024. "SLCO1B1 Genetic Variation Influence on Atorvastatin Systemic Exposure in Pediatric Hypercholesterolemia" Genes 15, no. 1: 99. https://doi.org/10.3390/genes15010099