
DNAJB11 Mutation in ADPKD Patients: Clinical Characteristics in a Monocentric Cohort
 , , , , , , , and
, , , , , , , and
Abstract
:1. Introduction
2. Materials and Methods
3. Results
3.1. Study Participants and Clinical Analysis
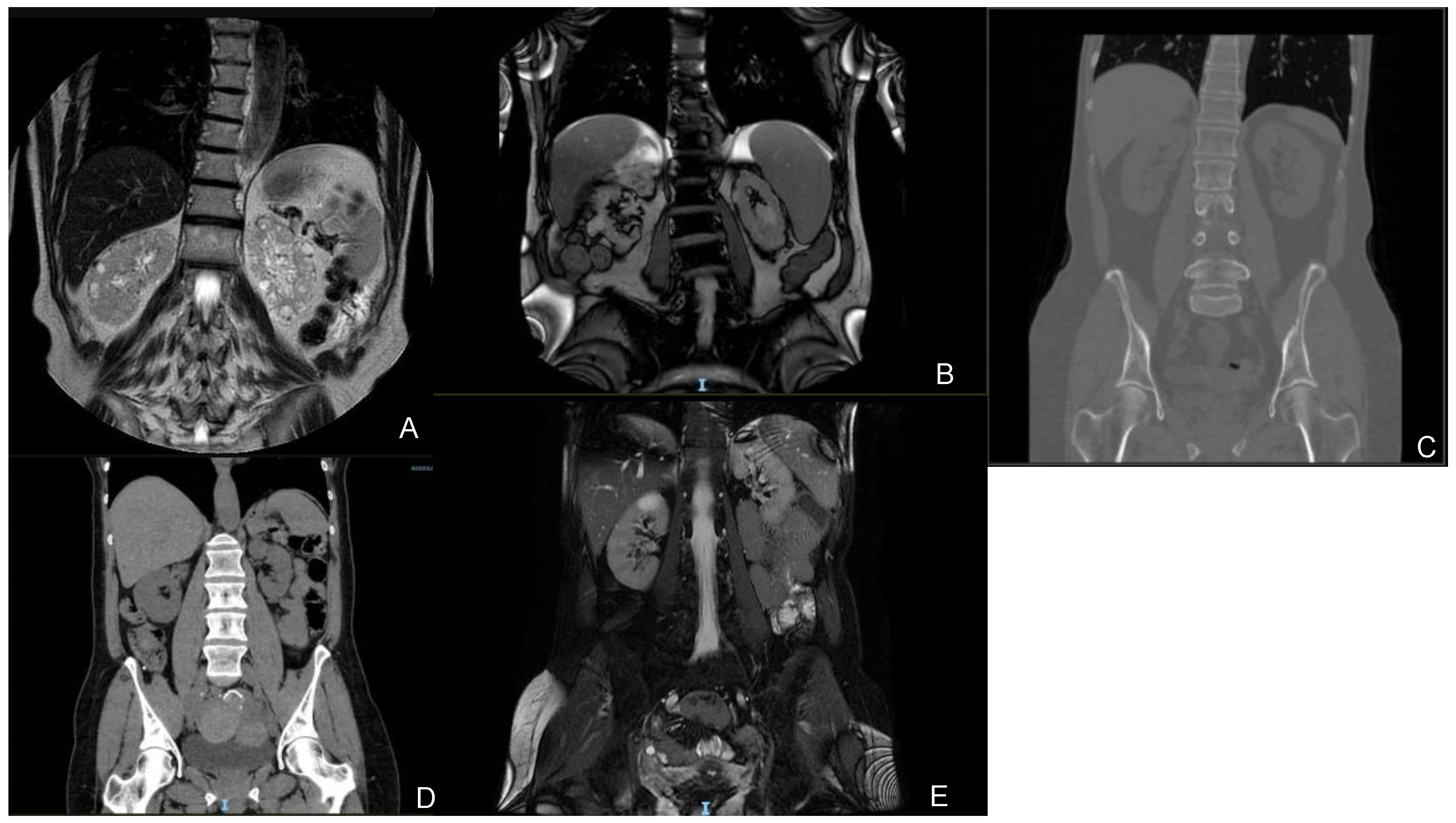
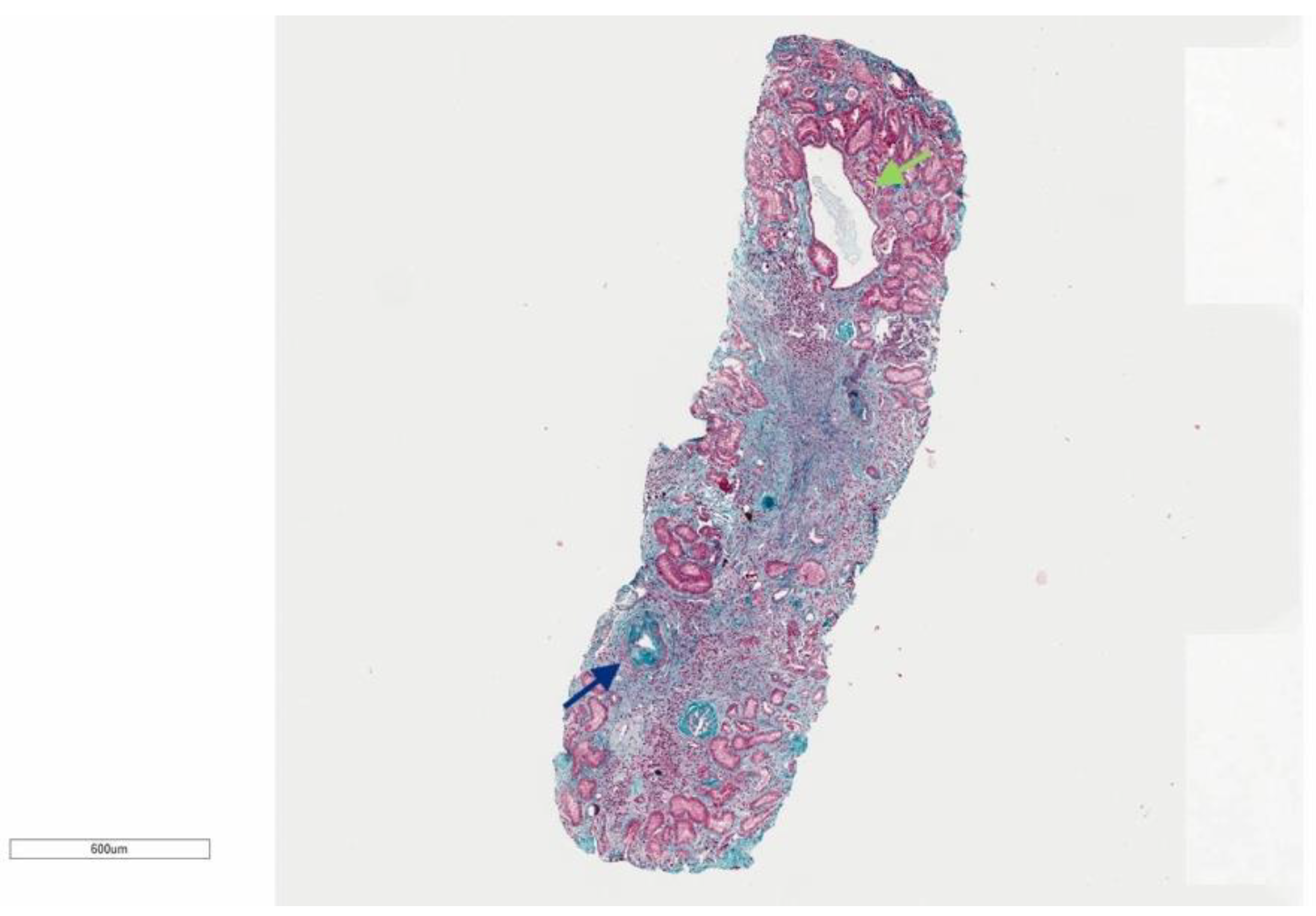
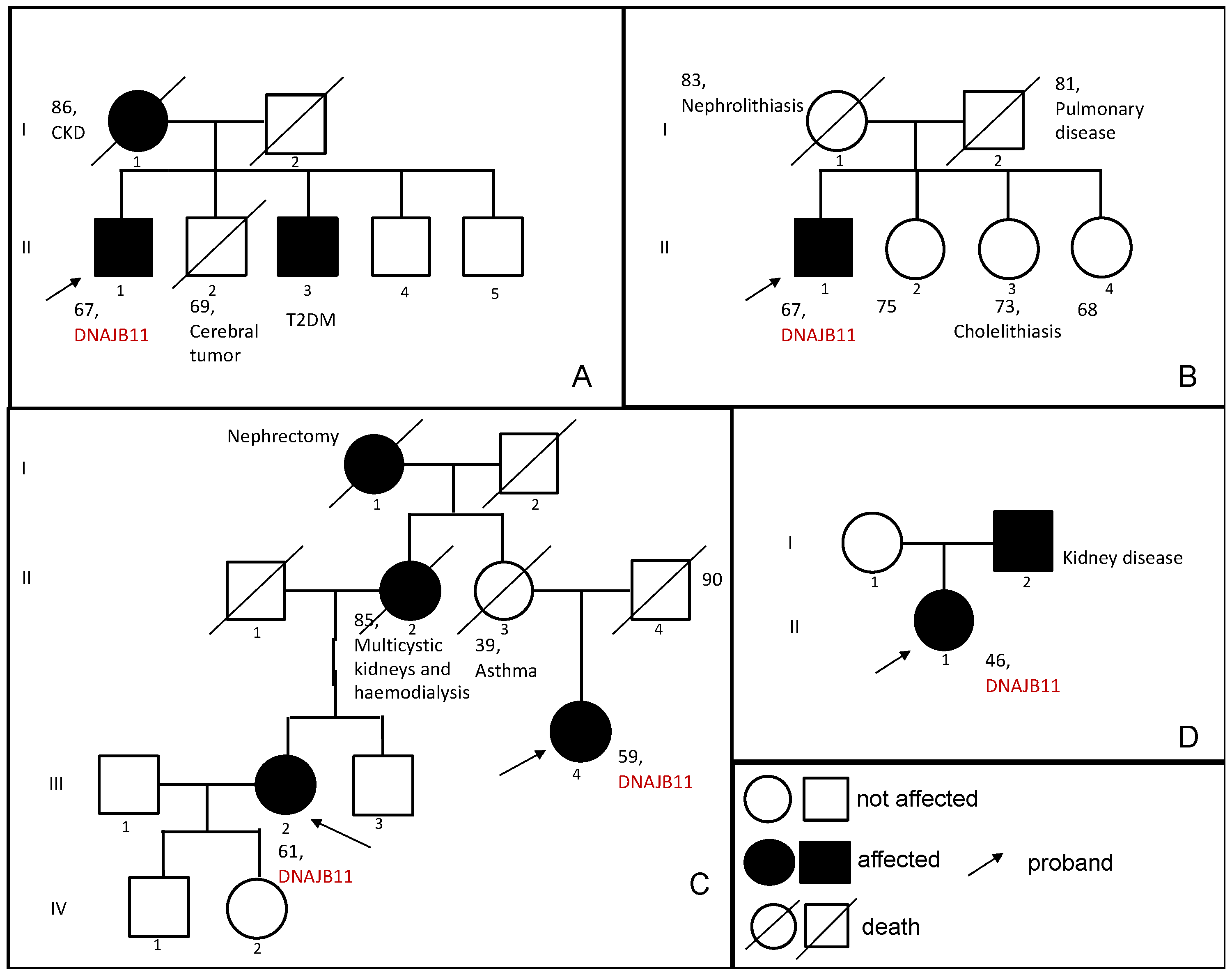
3.1.1. Family 1
3.1.2. Family 2
3.1.3. Family 3
3.1.4. Family 4
4. Discussion
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bergmann, C.; Guay-Woodford, L.M.; Harris, P.C.; Horie, S.; Peters, D.J.M.; Torres, V.E. Polycystic kidney disease. Nat. Rev. Dis. Primers 2018, 4, 50. [Google Scholar] [CrossRef]
- Colbert, G.B.; Elrggal, M.E.; Gaur, L.; Lerma, E.V. Update and review of adult polycystic kidney disease. Dis. Mon. 2020, 66, 100887. [Google Scholar] [CrossRef] [PubMed]
- Cornec-Le Gall, E.; Torres, V.E.; Harris, P.C. Genetic Complexity of Autosomal Dominant Polycystic Kidney and Liver Diseases. J. Am. Soc. Nephrol. 2018, 29, 13–23. [Google Scholar] [CrossRef]
- Gall, E.C.-L.; Olson, R.J.; Besse, W.; Heyer, C.M.; Gainullin, V.G.; Smith, J.M.; Audrézet, M.-P.; Hopp, K.; Porath, B.; Shi, B.; et al. Monoallelic Mutations to DNAJB11 Cause Atypical Autosomal-Dominant Polycystic Kidney Disease. Am. J. Hum. Genet. 2018, 102, 832–844. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Pei, Y.; Watnick, T. Diagnosis and screening of autosomal dominant polycystic kidney disease. Adv. Chronic Kidney Dis. 2010, 17, 140–152. [Google Scholar] [CrossRef] [PubMed]
- Irazabal, M.V.; Rangel, L.J.; Bergstralh, E.J.; Osborn, S.L.; Harmon, A.J.; Sundsbak, J.L.; Bae, K.T.; Chapman, A.B.; Grantham, J.J.; Mrug, M.; et al. Imaging classification of autosomal dominant polycystic kidney disease: A simple model for selecting patients for clinical trials. J. Am. Soc. Nephrol. 2015, 26, 160–172. [Google Scholar] [CrossRef] [PubMed]
- Cornec-Le Gall, E.; Alam, A.; Perrone, R.D. Autosomal dominant polycystic kidney disease. Lancet 2019, 393, 919–935. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Hendershot, L.M. ERdj3, a stress-inducible endoplasmic reticulum DnaJ homologue, serves as a cofactor for BiP’s interactions with unfolded substrates. Mol. Biol. Cell 2005, 16, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Huynh, V.T.; Audrézet, M.-P.; Sayer, J.A.; Ong, A.C.; Lefevre, S.; Le Brun, V.; Després, A.; Senum, S.R.; Chebib, F.T.; Barroso-Gil, M.; et al. Clinical spectrum, prognosis and estimated prevalence of DNAJB11-kidney disease. Kidney Int. 2020, 98, 476–487. [Google Scholar] [CrossRef] [PubMed]
- Fedeles, S.V.; Gallagher, A.R.; Somlo, S. Polycystin-1: A master regulator of intersecting cystic pathways. Trends Mol. Med. 2014, 20, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Fedeles, S.V.; Tian, X.; Gallagher, A.-R.; Mitobe, M.; Nishio, S.; Lee, S.H.; Cai, Y.; Geng, L.; Crews, C.M.; Somlo, S. A genetic interaction network of five genes for human polycystic kidney and liver diseases defines polycystin-1 as the central determinant of cyst formation. Nat. Genet. 2011, 43, 639–647. [Google Scholar] [CrossRef] [PubMed]
- Besse, W.; Dong, K.; Choi, J.; Punia, S.; Fedeles, S.V.; Choi, M.; Gallagher, A.-R.; Huang, E.B.; Gulati, A.; Knight, J.; et al. Isolated polycystic liver disease genes define effectors of polycystin-1 function. J. Clin. Investig. 2017, 127, 3558. [Google Scholar] [CrossRef] [PubMed]
- Wilson, G.J.; Wood, S.; Patel, C.; Oliver, K.; John, G.; Ranganathan, D.; Mallett, A.; Isbel, N. DNAJB11-Related Atypical ADPKD in a Kidney Transplant Donor. Kidney Int. Rep. 2020, 5, 1363–1366. [Google Scholar] [CrossRef] [PubMed]
- Pisani, I.; Allinovi, M.; Palazzo, V.; Zanelli, P.; Gentile, M.; Farina, M.T.; Giuliotti, S.; Cravedi, P.; Delsante, M.; Maggiore, U.; et al. More dissimilarities than affinities between DNAJB11-PKD and ADPKD. Clin. Kidney J. 2022, 15, 1179–1187. [Google Scholar] [CrossRef] [PubMed]
- Gall, E.C.-L.; Audrézet, M.-P.; Rousseau, A.; Hourmant, M.; Renaudineau, E.; Charasse, C.; Morin, M.-P.; Moal, M.-C.; Dantal, J.; Wehbe, B.; et al. The PROPKD Score: A New Algorithm to Predict Renal Survival in Autosomal Dominant Polycystic Kidney Disease. J. Am. Soc. Nephrol. 2016, 27, 942–951. [Google Scholar] [CrossRef] [PubMed]
- Müller, R.-U.; Messchendorp, A.L.; Birn, H.; Capasso, G.; Gall, E.C.-L.; Devuyst, O.; van Eerde, A.; Guirchoun, P.; Harris, T.; Hoorn, E.J.; et al. An update on the use of tolvaptan for autosomal dominant polycystic kidney disease: Consensus statement on behalf of the ERA Working Group on Inherited Kidney Disorders, the European Rare Kidney Disease Reference Network and Polycystic Kidney Disease International. Nephrol. Dial. Transplant. 2022, 37, 825–839. [Google Scholar] [PubMed]
- Roy, S.G.; Li, Z.; Guo, Z.; Long, K.T.; Rehrl, S.; Tian, X.; Dong, K.; Besse, W. Dnajb11-Kidney Disease Develops from Reduced Polycystin-1 Dosage but not Unfolded Protein Response in Mice. J. Am. Soc. Nephrol. 2023, 34, 1521–1534. [Google Scholar]
- Mariniello, M.; Schiano, G.; Yoshifuji, A.; Gillion, V.; Sayer, J.A.; Jouret, F.; Le Meur, Y.; Gall, E.C.-L.; Olinger, E.G.; Devuyst, O. Uromodulin Processing in DNAJB11-kidney Disease. Kidney Int. 2023. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| DNAJB11 Mutation | Kidney Imaging (Diameter in cm of Right/Left Kidney) | Liver Imaging | Hypertension (age) | Renal Function at Diagnosis | Renal Function at Last Follow-Up | Δ eGFR per year | Other Conditions | |
|---|---|---|---|---|---|---|---|---|
| Patient 1 (67) | NM_016306.5:c.716T>G p.(Leu239Ter) 4 / | Regular-sized kidneys (10/10) Bilateral cortical millimetric cysts | No cysts | Yes (67) | Creat. 1.58 mg/dL eGFR 46 mL/min/1.73 m2 CKD IIIa | Creat. 2.50 mg/dL eGFR 26 mL/min/1.73 m2 CKD IV | −4 | MGUS, mild bilateral perceptive hearing loss, hyperuricemia |
| Patient 2 (67) | NM_016306.5:c.134A>G p.(Tyr45Cys) 4 gnomAD Exomes: Version: 2.1.1ƒ = 0.00000797; gnomAD GenomesVersion: 2.1.1ƒ = 0.0000319 | Regular-sized kidneys (11/13) Bilateral millimetric cysts and some bigger cysts | No cysts | Yes | Creat. 2.45 mg/dL eGFR 27 mL/min/1.73 m2 CKD IV | Creat. 4.16 mg/dL eGFR 14 mL/min/1.73 m2 CKD V | −4.3 | Nephrolithiasis, AMI, AF, severe mitral insufficiency |
| Patient 3 (61) | NM_016306.5:c.456+3_456+6del (IVS4) 4 / | Regular-sized kidneys (10.9/10.5) Bilateral millimetric cysts | No cysts | Yes (55) | Creat. 1.04 mg/dL eGFR 59 mL/min/1.73 m2 CKD IIIa | Creat. 1.14 mg/dL eGFR 52 mL/min/1.73 m2 CKD IIIa | −2.3 | Microhaematuria, recurrent cystitis, nephrolithiasis, mild tricuspid insufficiency, mild neurosensorial hearing loss in the left ear |
| Patient 4 (59) | NM_016306.5:c.456+3_456+6del (IVS4) 4 / | Regular-sized kidneys (10.3/9.4) Bilateral millimetric cysts | Multiple cysts | Yes (50) | Creat. 1.27 mg/dL eGFR 46 mL/min/1.73 m2 CKD IIIa | Creat. 1.27 mg/dL eGFR 46 mL/min/1.73 m2 CKD IIIa | N/A | / |
| Patient 5 (46) | NM_016306.5:c.499C>T p.(Arg167Trp) 3 gnomAD ExomesVersion: 2.1.1ƒ = 0.00000398; gnomAD GenomesVersion: 2.1.1 / | Regular-sized kidneys (10/10) Bilateral millimetric cysts | One cyst in the III segment (4 mm) | No | Creat. 1.54 mg/dL eGFR 40 mL/min/1.73 m2 CKD IIIa | Creat. 1.54 mg/dL eGFR 40 mL/min/1.73 m2 CKD IIIa | N/A | Hyperuricemia, reactive anxiety depressive syndrome |
| Kidney Imaging | Liver Imaging | Hypertension | Laboratory | Other Conditions | |||
|---|---|---|---|---|---|---|---|
| Presence of cysts | Mean diameter | CKD | ESRD | ||||
| Cornec- Le Gall et al. [4] | MBSC: 78.3% Unilateral cysts: 8.7% No cysts: 4.3% N/A: 8.7% | 10.3 ± 1.7 cm N/A: 13.0% | Cysts: 39.1% No cysts: 47.8% N/A: 13.1% | 47.8% 56.3 ± 8.4 years | 69.6% 53.4 ± 17.3 years eGFR: 75.1 ± 25.3 mL/min/1.73 m2 | 30.4% 77 ± 10.8 years | Nephrolithiasis: 8.7% T2DM: 8.7% Hyperuricemia: 13.0% |
| Huynh et al. [10] | MBSC: 83.3% Polycystic kidneys: 5.6% No cysts: 1.8% N/A: 9.3% | 12.2 ± 2.2 cm N/A: 35.2% | Cysts: 35.2% No cysts: 40.7% N/A: 24.1% | 57.4% 55.1 ± 12.1 years | 55.6% 58.1 ± 11.1 years eGFR: 52.7 ± 31.6 mL/min/1.73 m2 | 44.4% 71.6 ± 6.8 years | T2DM: 11.1% Hyperuricemia: 5.6% ICA: 3.7% Thoracic aortic aneurysms: 3.7% |
| Pisani et al. [15] | Cysts: 81.5% No cysts: 18.5% | 10.3 ± 1.6 cm N/A: 18.5% | Cysts: 40.7% No cysts: 44.5% N/A: 14.8% | 59.3% | N/A | 44.4% 71.1 ± 4.5 years | Nephrolithiasis: 59.3% T2DM: 18.5% Cardiac valvular defects: 3.7% |
| Bologna cohort | MBSC: 100.0% No cysts: 0.0% N/A: 0.0% | 10.7 ± 1.2 cm N/A: 0.0% | Cysts: 40.0% No cysts: 60.0% N/A: 0.0% | 80.0% 57.3 ± 8.7 years | 100.0% 60.0 ± 8.6 years eGFR: 35.6 ± 15.5 mL/min/1.73 m2 | 0.0 % | Nephrolithiasis: 40.0% Hyperuricemia: 40.0% Cardiac valvular defects: 40.0% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aiello, V.; Ciurli, F.; Conti, A.; Cristalli, C.P.; Lerario, S.; Montanari, F.; Sciascia, N.; Vischini, G.; Fabbrizio, B.; Di Costanzo, R.; et al. DNAJB11 Mutation in ADPKD Patients: Clinical Characteristics in a Monocentric Cohort. Genes 2024, 15, 3. https://doi.org/10.3390/genes15010003
Aiello V, Ciurli F, Conti A, Cristalli CP, Lerario S, Montanari F, Sciascia N, Vischini G, Fabbrizio B, Di Costanzo R, et al. DNAJB11 Mutation in ADPKD Patients: Clinical Characteristics in a Monocentric Cohort. Genes. 2024; 15(1):3. https://doi.org/10.3390/genes15010003
Chicago/Turabian StyleAiello, Valeria, Francesca Ciurli, Amalia Conti, Carlotta Pia Cristalli, Sarah Lerario, Francesca Montanari, Nicola Sciascia, Gisella Vischini, Benedetta Fabbrizio, Roberta Di Costanzo, and et al. 2024. "DNAJB11 Mutation in ADPKD Patients: Clinical Characteristics in a Monocentric Cohort" Genes 15, no. 1: 3. https://doi.org/10.3390/genes15010003