
Molecular Characterization and Genome Mechanical Features of Two Newly Isolated Polyvalent Bacteriophages Infecting Pseudomonas syringae pv. garcae
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Biological Material
2.2. Chemicals
2.3. Phage PEG-Precipitation
2.4. Transmission Electron Microscopy (TEM) Analyses
2.5. Phage Virion Whole Genome Sequencing
2.6. Phage Virion Genome Assembly, Taxonomic Evaluation, Annotation, and Phylogeny
2.7. Mechanical Properties of the PsgM02F and PsgM04F Phage Genomes
2.8. Viral Proteomic Trees
2.9. Statistical Analyses
3. Results
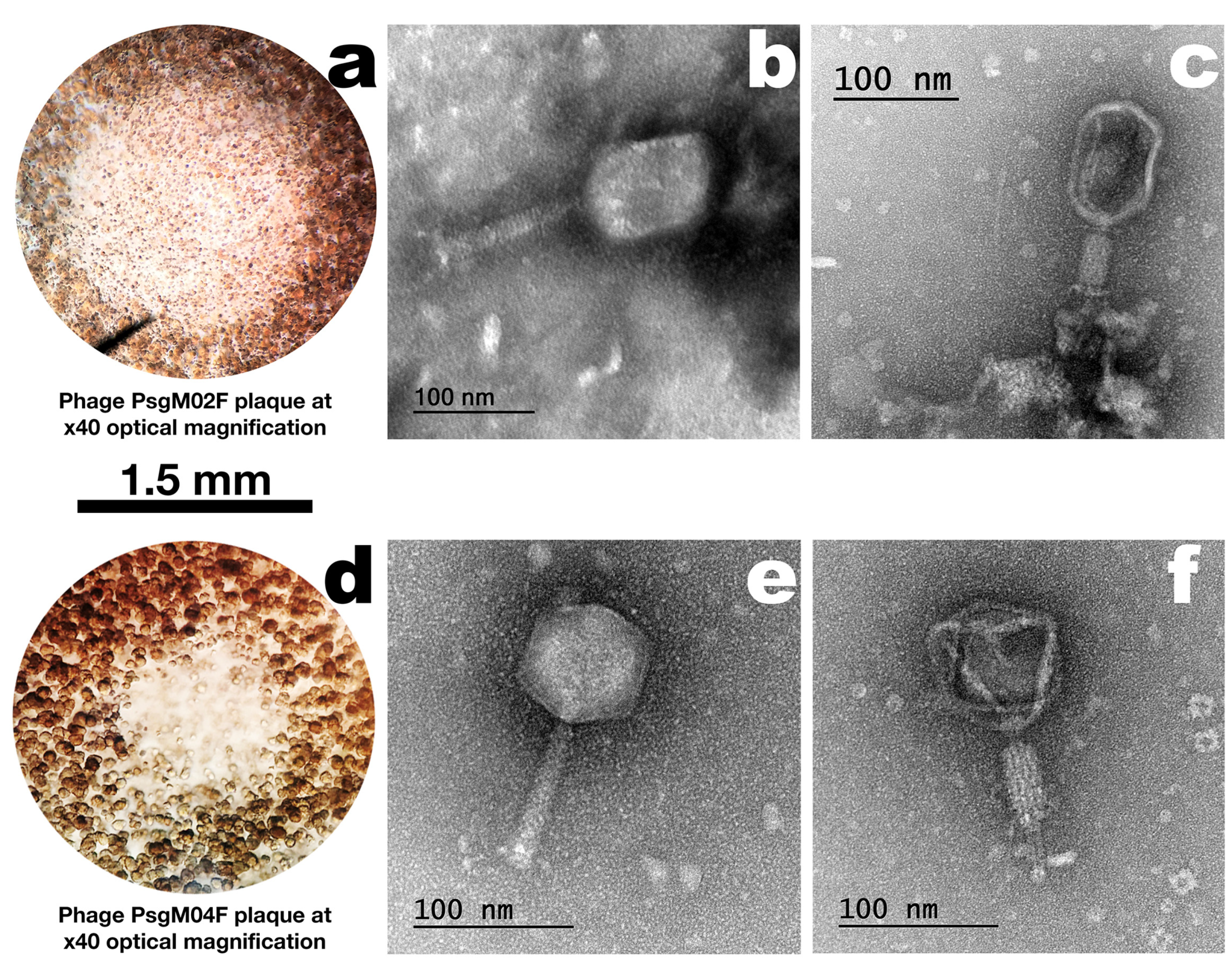
3.1. Phage Plaque Morphologies and Virion Morphotypes and Physical Features
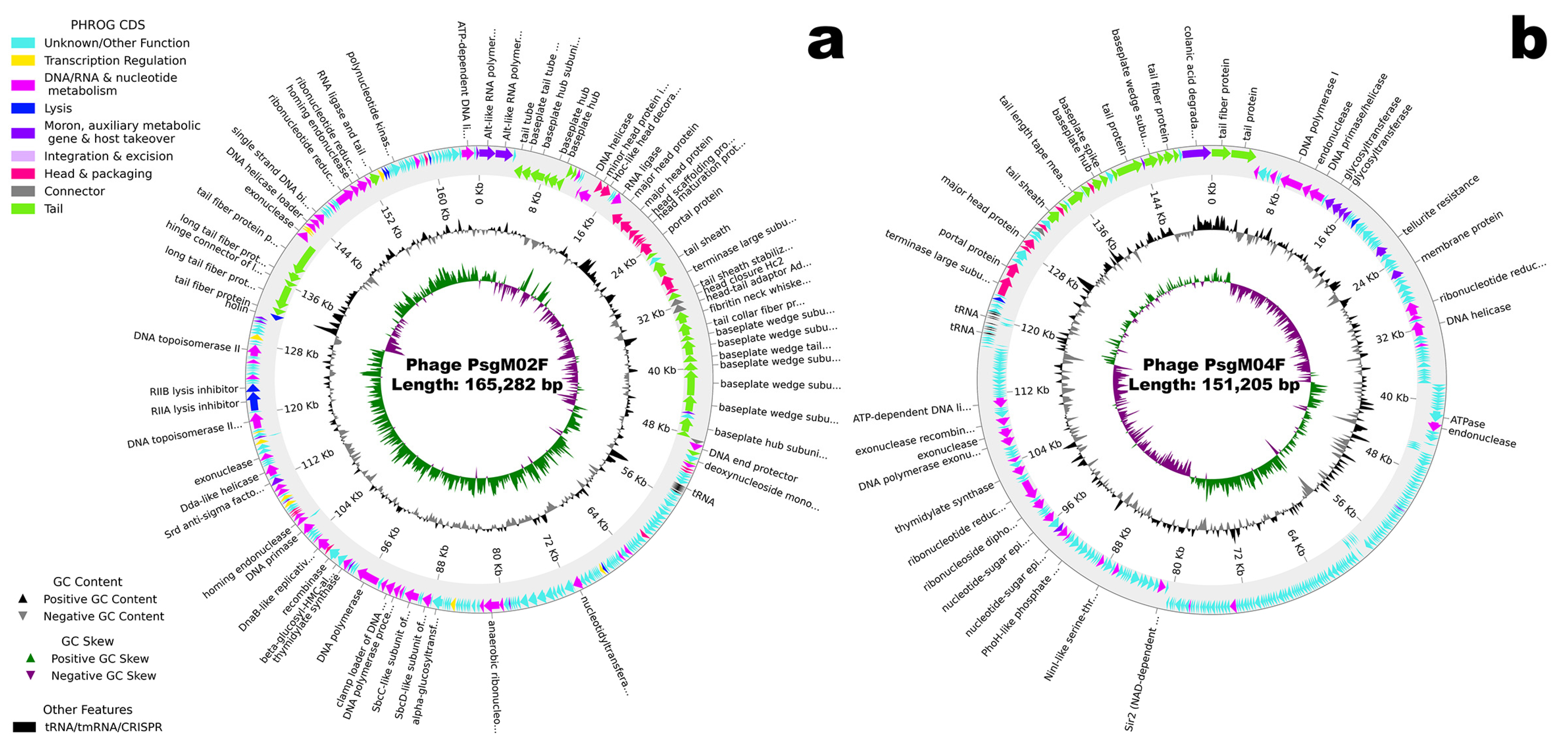
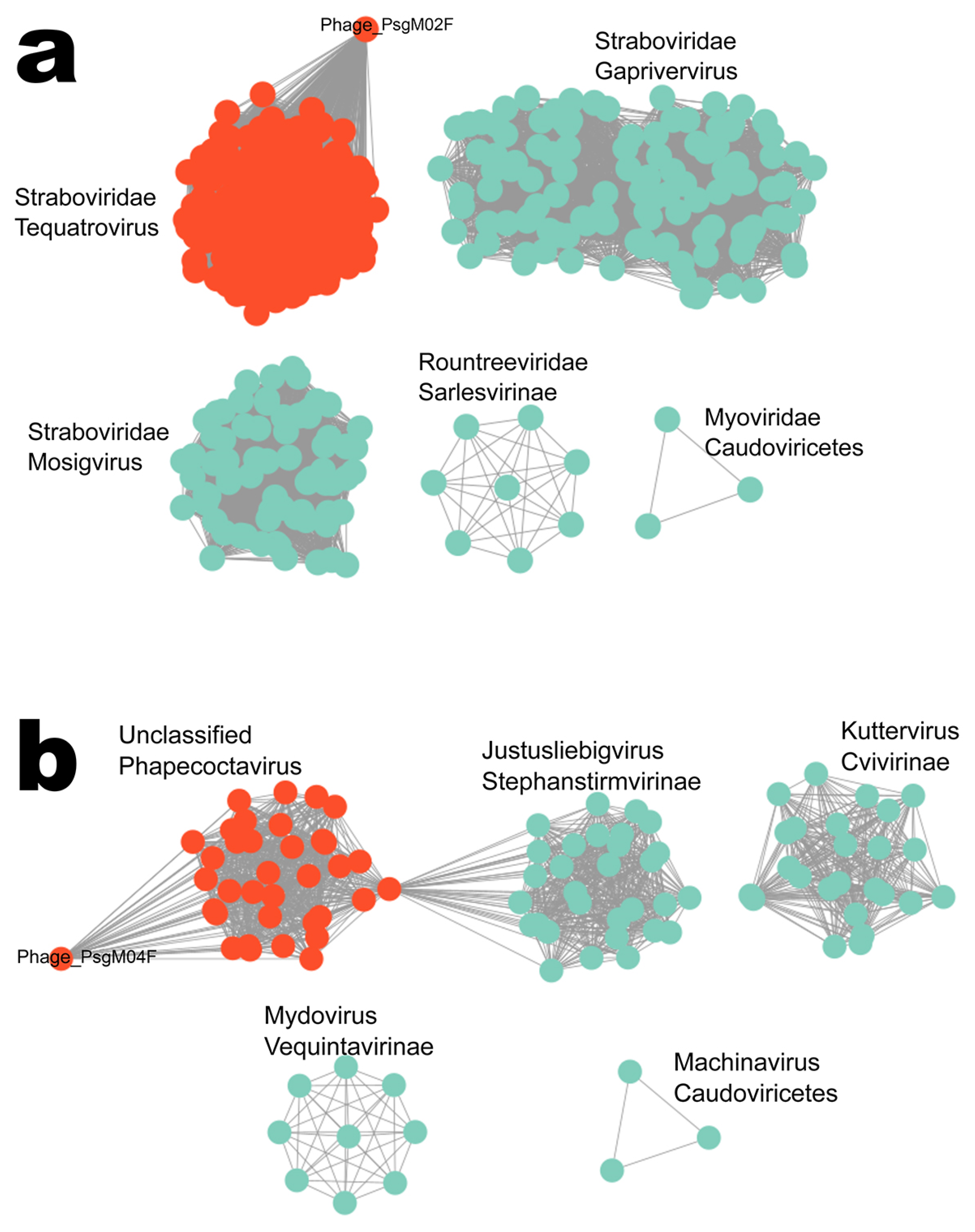
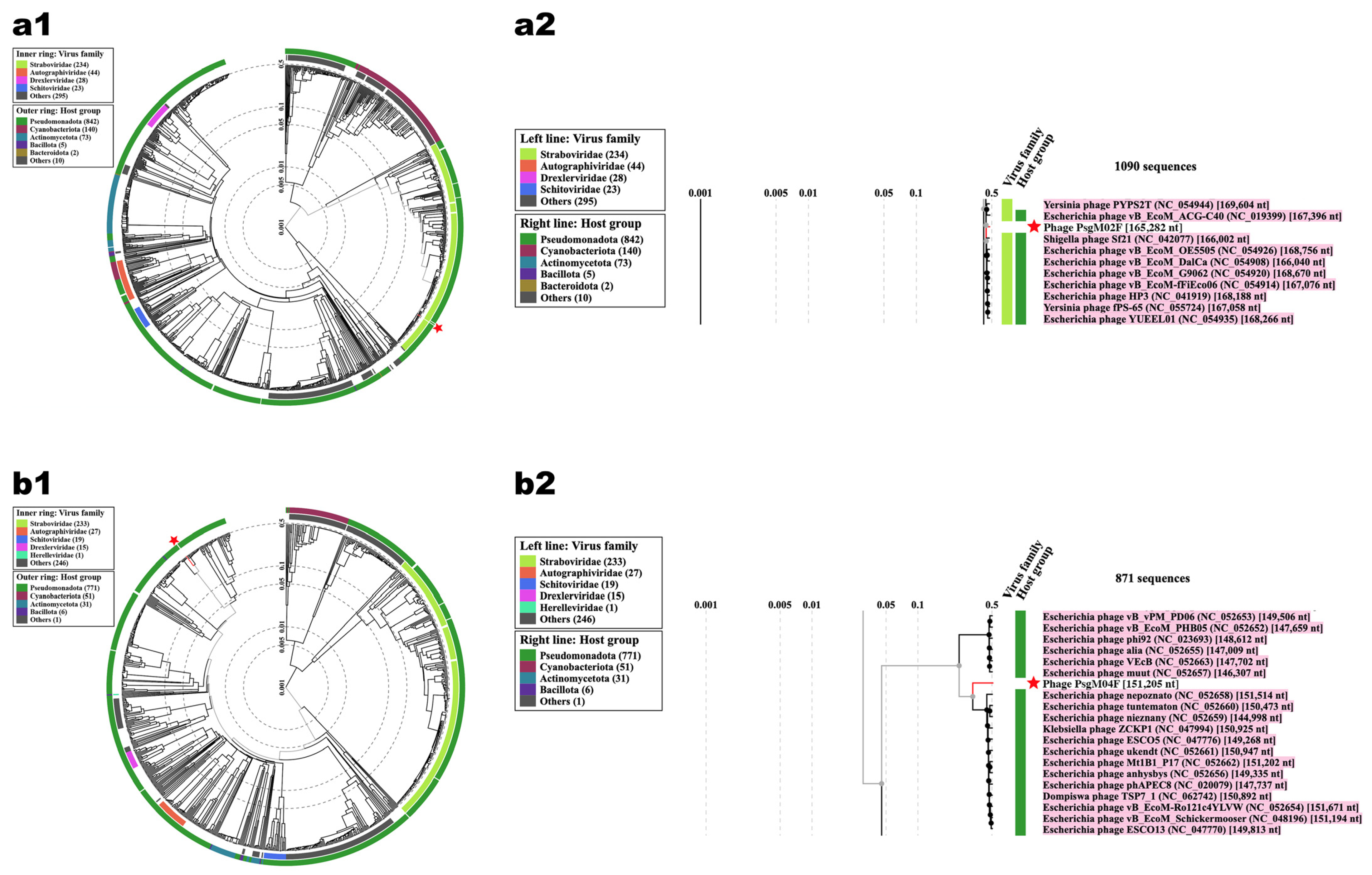
3.2. Genomic Characterization
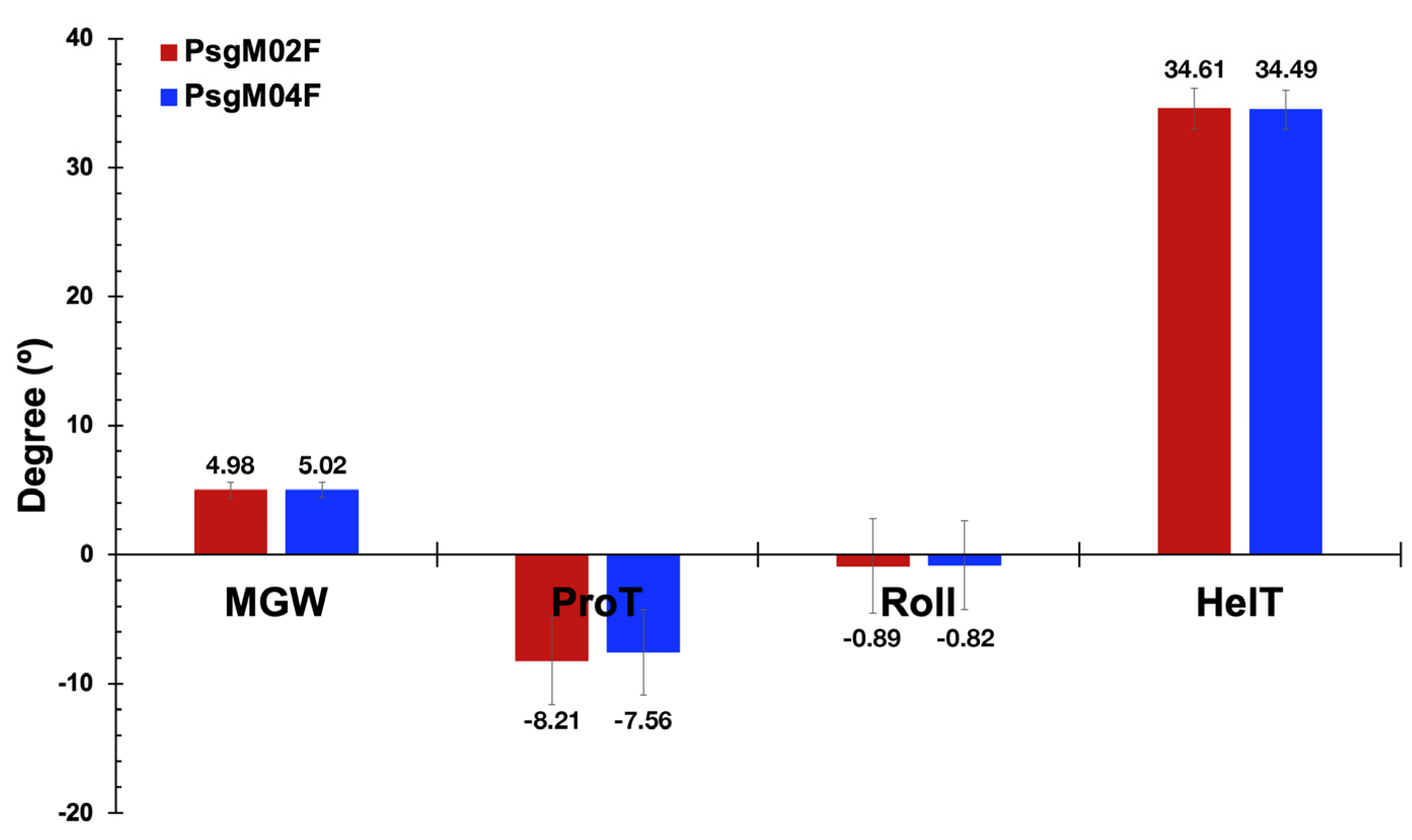
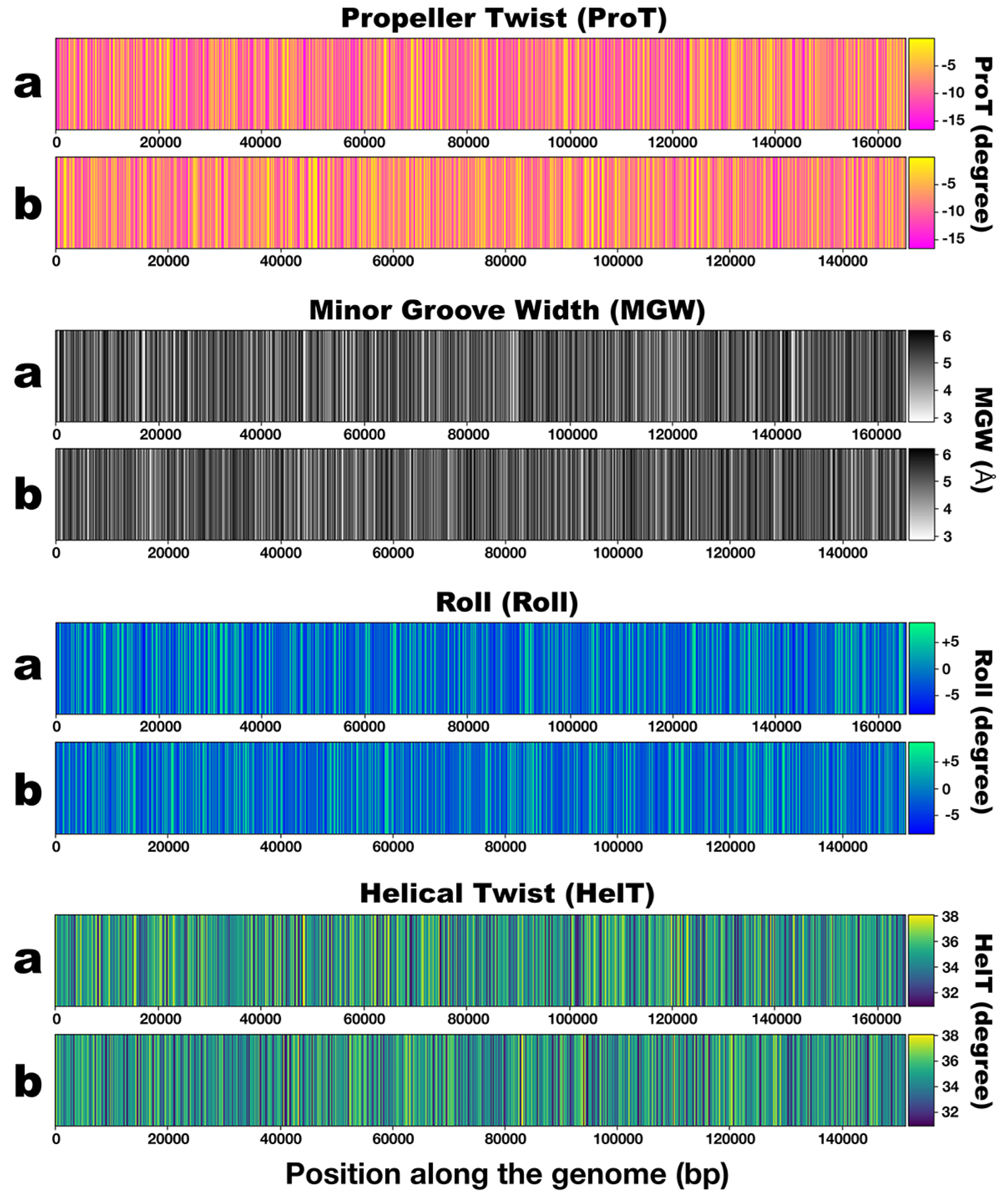
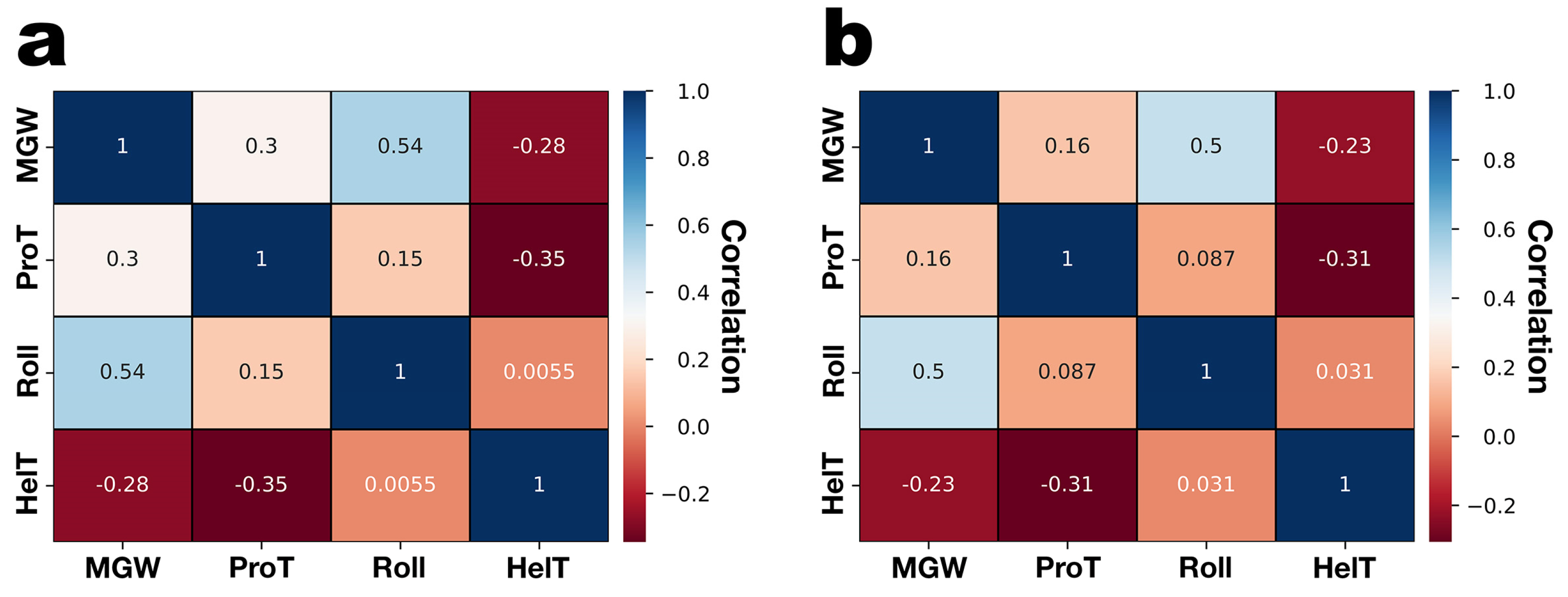
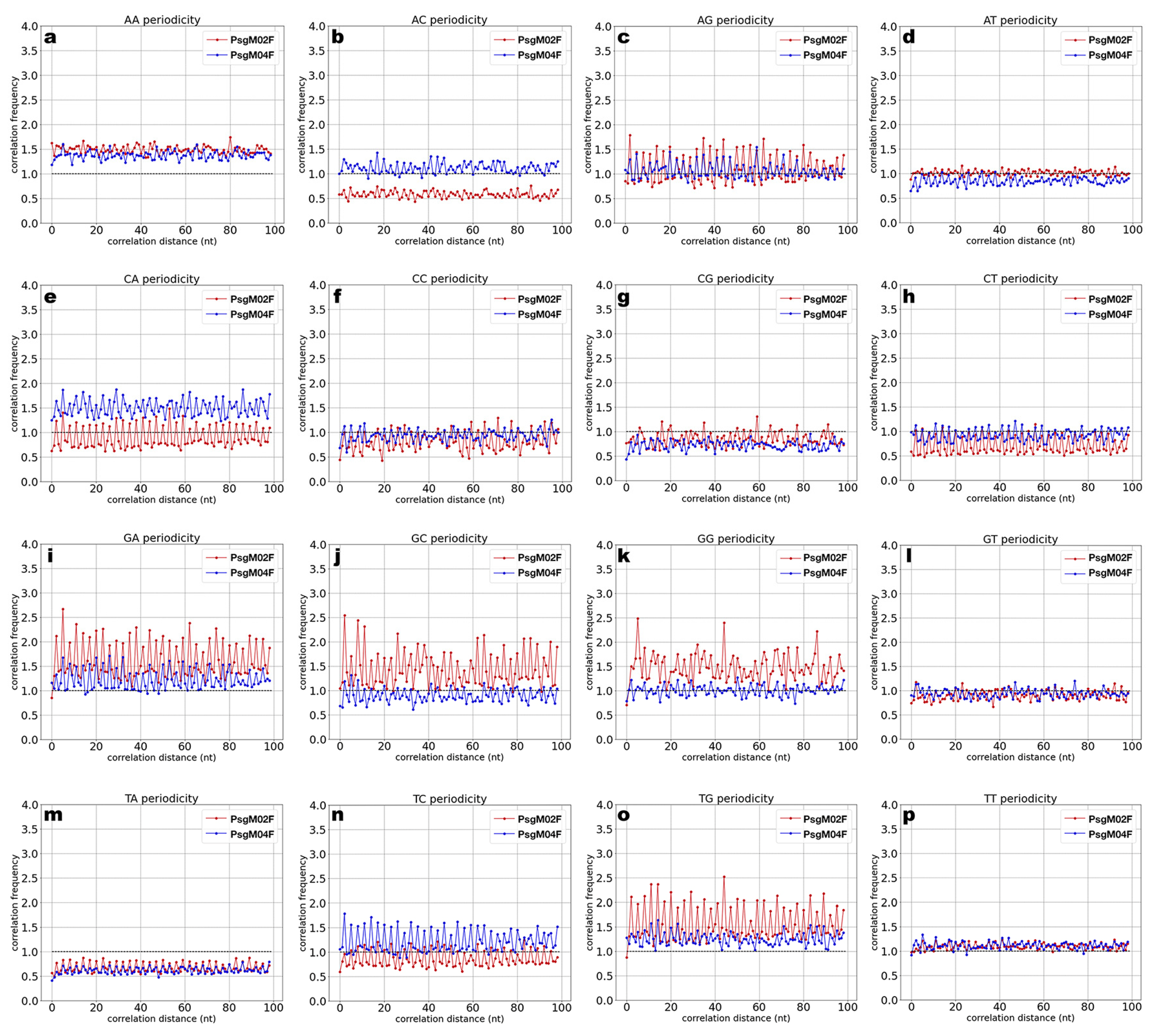
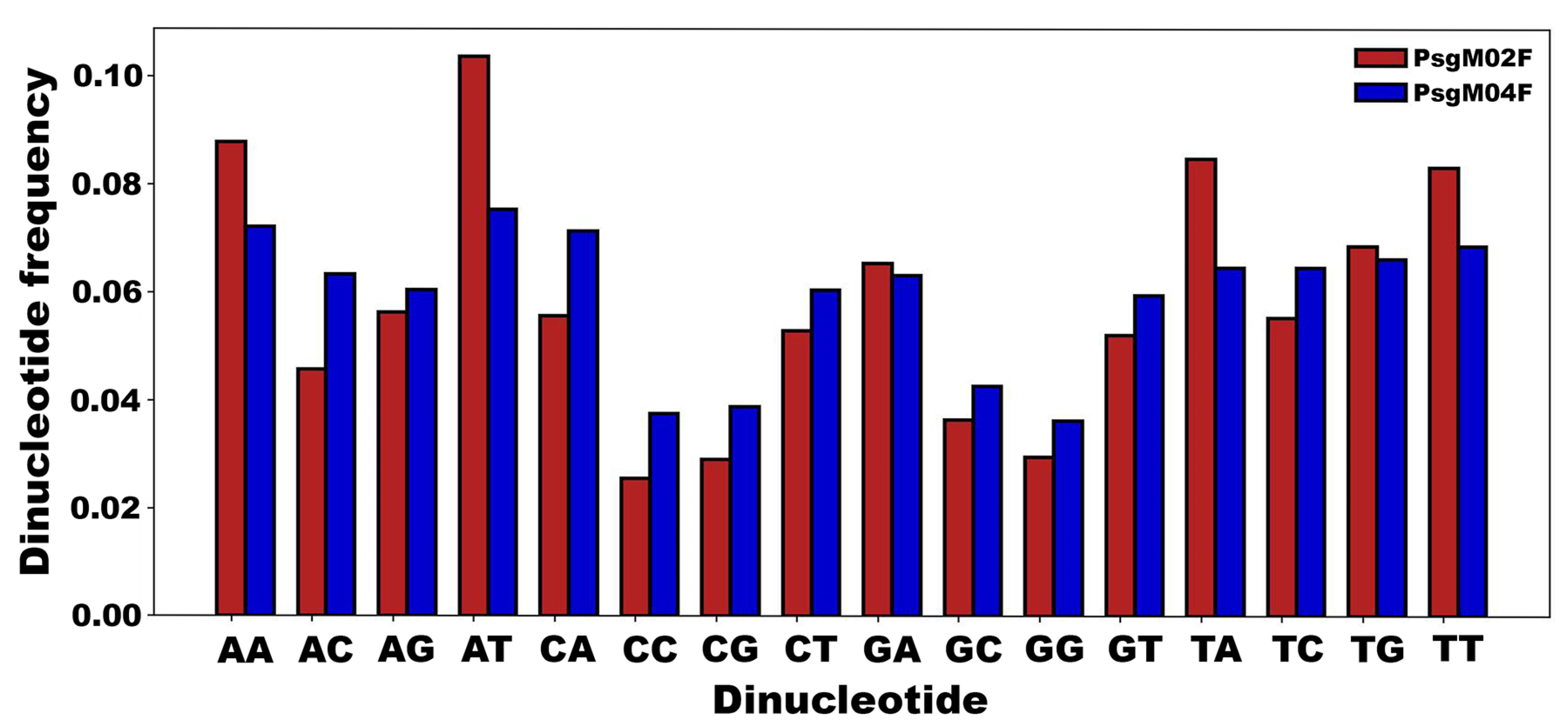
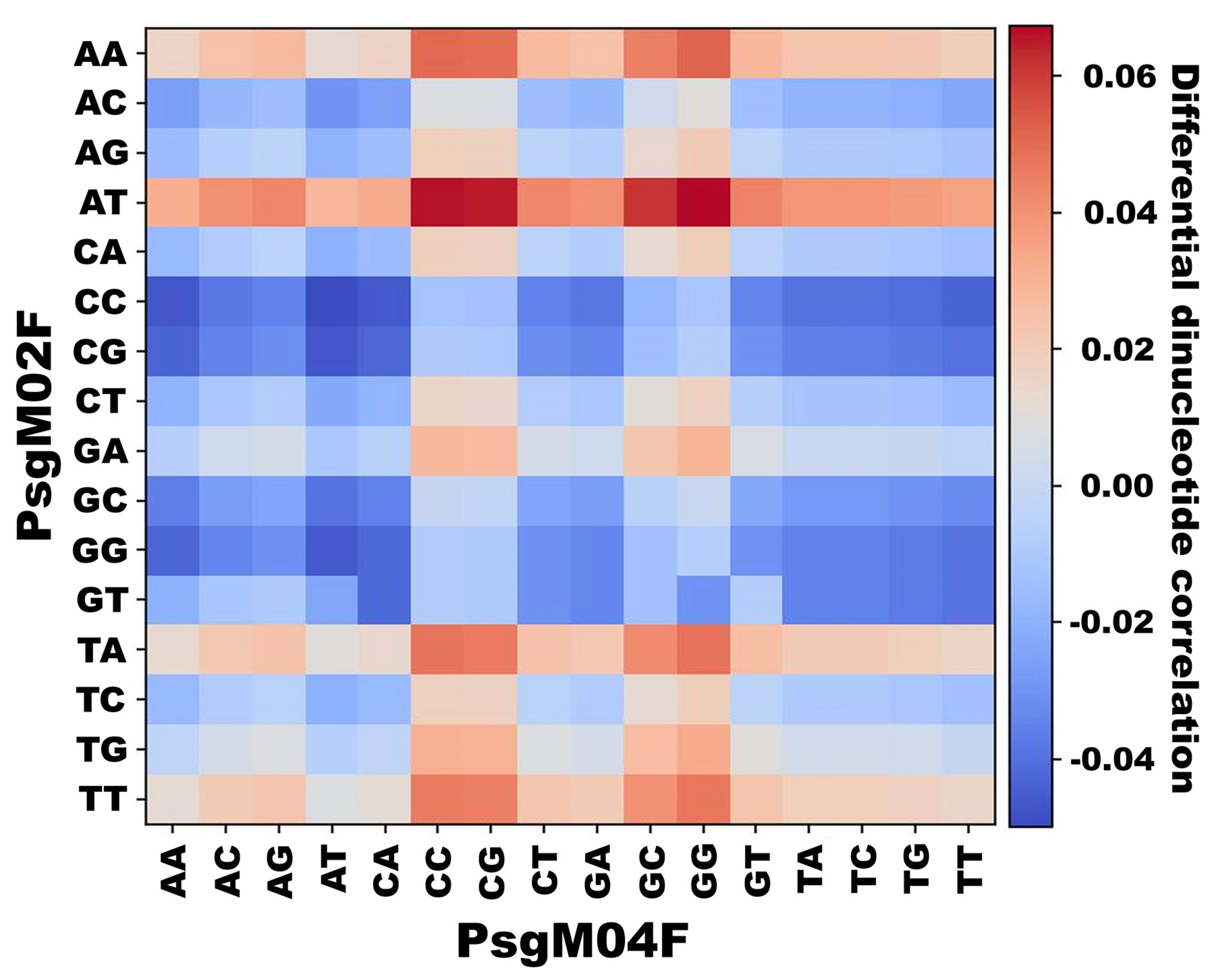
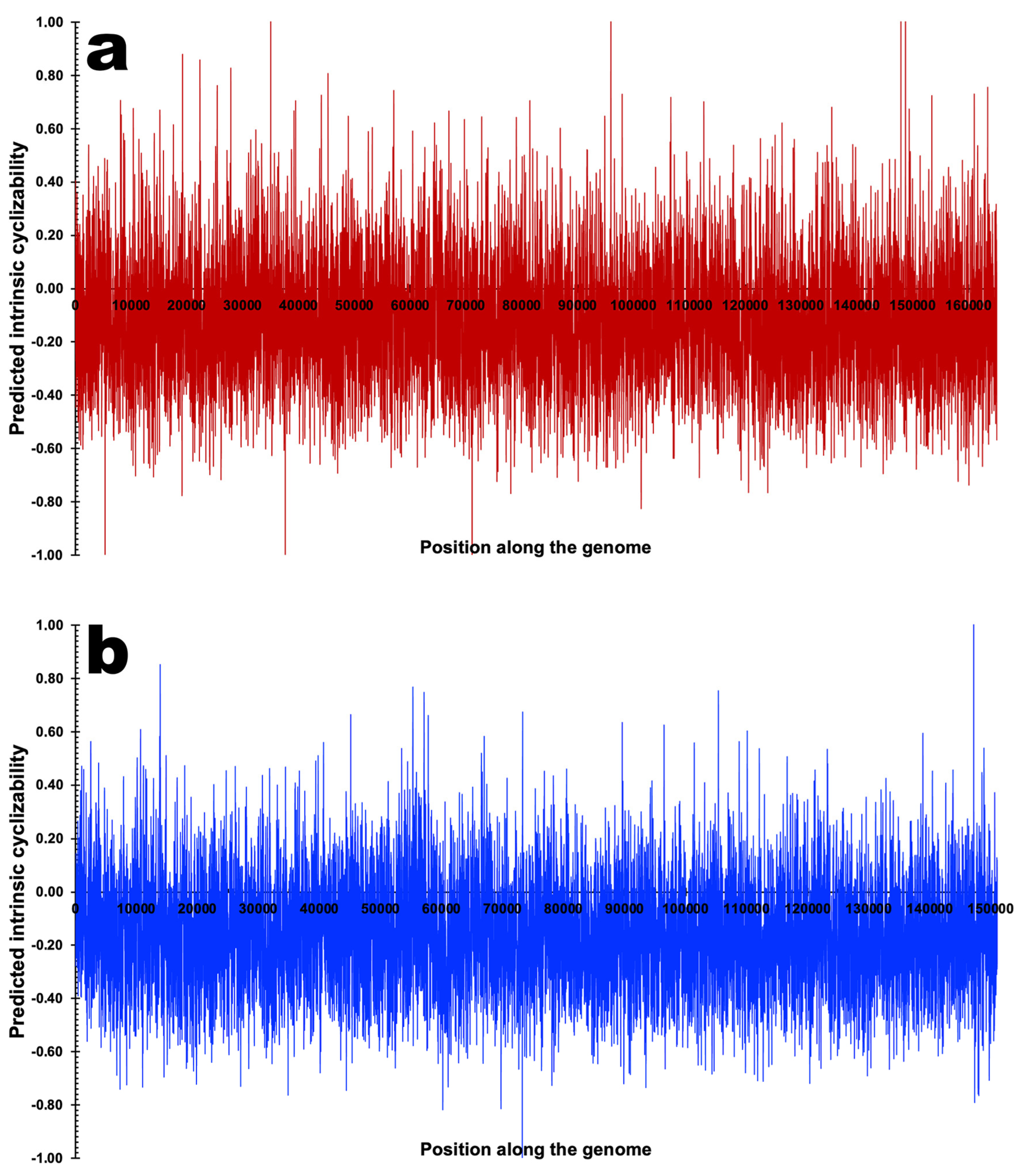
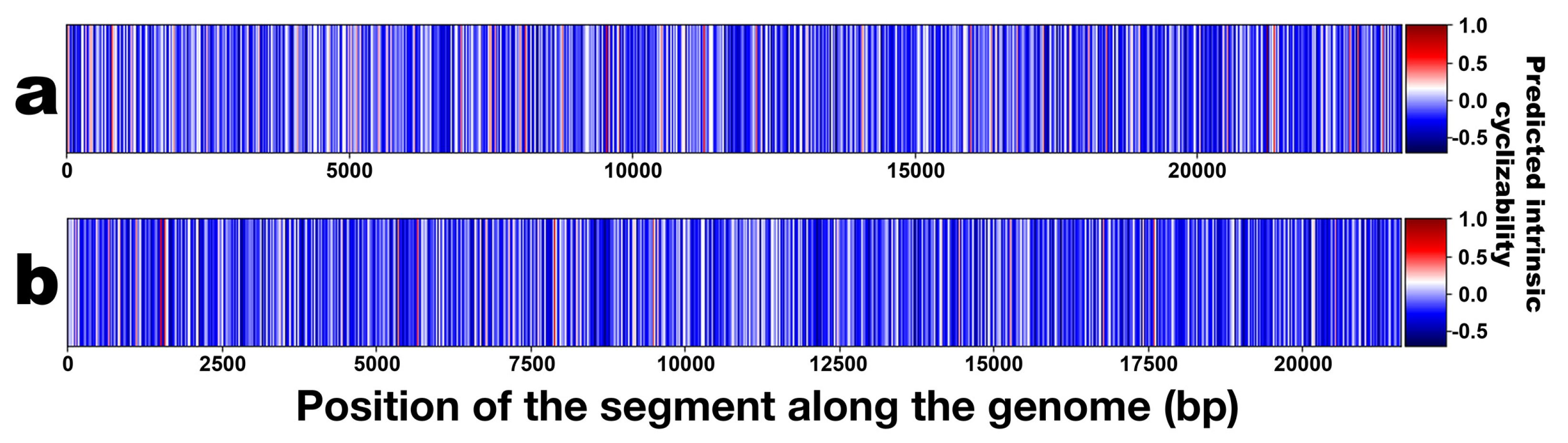
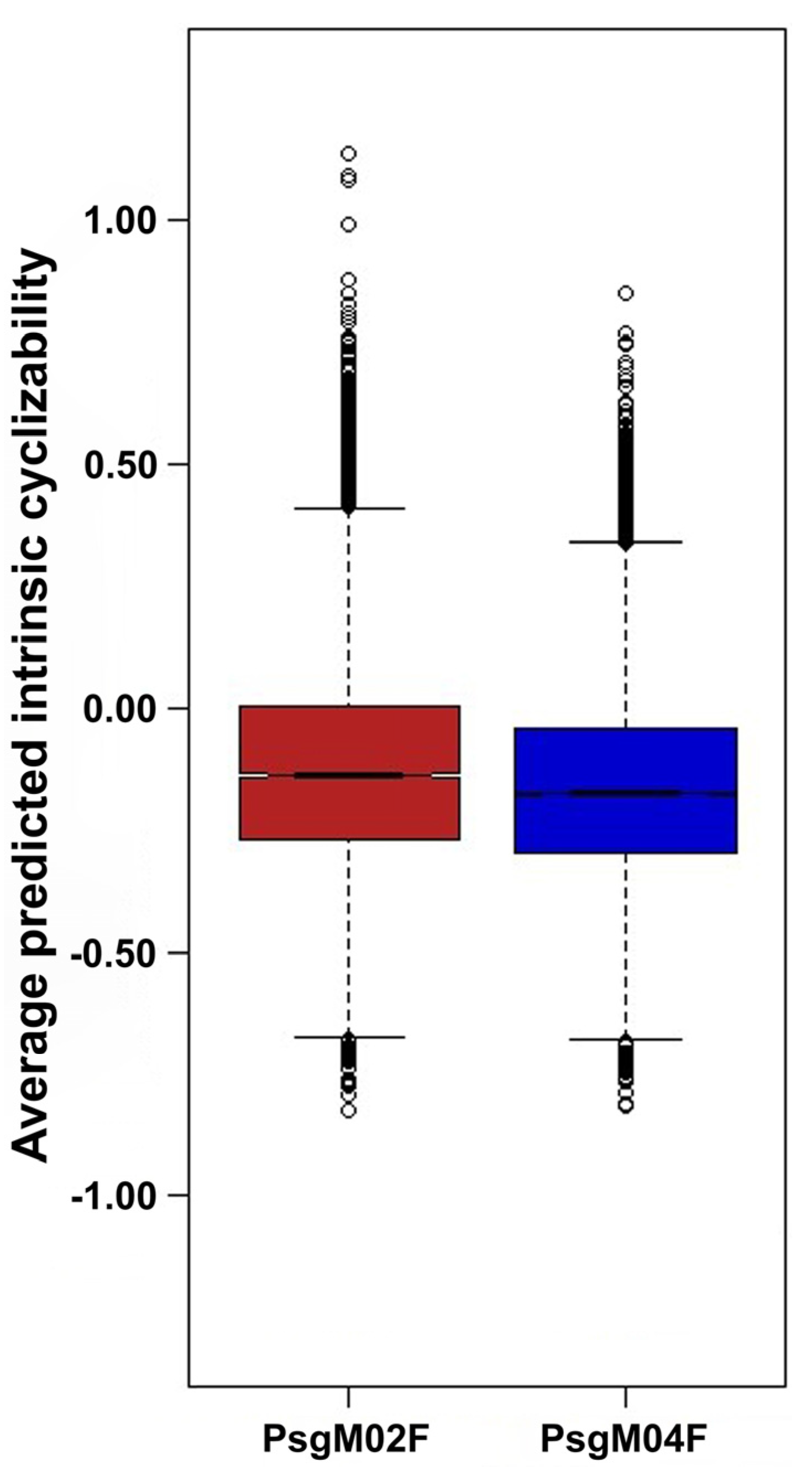
3.3. DNA Structural Features of Phages PsgM02F and PsgM04F
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Savary, S.; Willocquet, L.; Pethybridge, S.J.; Esker, P.; McRoberts, N.; Nelson, A. The global burden of pathogens and pests on major food crops. Nat. Ecol. Evol. 2019, 3, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.K.; Delgado-Baquerizo, M.; Egidi, E.; Guirado, E.; Leach, J.E.; Liu, H.; Trivedi, P. Climate change impacts on plant pathogens, food security and paths forward. Nat. Rev. Microbiol. 2023, 21, 640–656. [Google Scholar] [CrossRef] [PubMed]
- Ristaino, J.B.; Anderson, P.K.; Bebber, D.P.; Brauman, K.A.; Cunniffe, N.J.; Fedoroff, N.V.; Finegold, C.; Garrett, K.A.; Gilligan, C.A.; Jones, C.M.; et al. The persistent threat of emerging plant disease pandemics to global food security. Proc. Natl. Acad. Sci. USA 2021, 118, e2022239118. [Google Scholar] [CrossRef]
- Savary, S.; Willocquet, L. Modeling the Impact of Crop Diseases on Global Food Security. Annu. Rev. Phytopathol. 2020, 58, 313–341. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Newton, A.C. Climate change, plant diseases and food security: An overview. Plant Pathol. 2011, 60, 2–14. [Google Scholar] [CrossRef]
- Rizzo, D.M.; Lichtveld, M.; Mazet, J.A.K.; Togami, E.; Miller, S.A. Plant health and its effects on food safety and security in a One Health framework: Four case studies. One Health Outlook 2021, 3, 6. [Google Scholar] [CrossRef]
- Yamada, J.K. Resistance of Pseudomonas syringae pv. garcae to copper [Resistência de isolados de Pseudomonas syringae pv. garcae ao cobre]. Master’s Thesis, Federal University of Lavras, Lavras, Brazil, 2014. [Google Scholar]
- Pereira, C.; Costa, P.; Pinheiro, L.; Balcão, V.M.; Almeida, A. Kiwifruit bacterial canker: An integrative view focused on biocontrol strategies. Planta 2021, 253, 49. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, L.A.M.; Pereira, C.; Frazão, C.; Balcão, V.M.; Almeida, A. Efficiency of Phage φ6 for Biocontrol of Pseudomonas syringae pv. syringae: An in Vitro Preliminary Study. Microorganisms 2019, 7, 286. [Google Scholar] [CrossRef]
- Pinheiro, L.A.M.; Pereira, C.; Barreal, M.E.; Gallego, P.P.; Balcão, V.M.; Almeida, A. Use of phage ϕ6 to inactivate Pseudomonas syringae pv. actinidiae in kiwifruit plants: In vitro and ex vivo experiments. Appl. Microbiol. Biotechnol. 2019, 104, 1319–1330. [Google Scholar] [CrossRef]
- McManus, P.S.; Stockwell, V.O.; Sundin, G.W.; Jones, A.L. Antibiotic use in plant agriculture. Annu. Rev. Phytopathol. 2002, 40, 443–465. [Google Scholar] [CrossRef]
- Silva, E.C.; Rodrigues, L.M.R.; Vila, M.M.D.C.; Balcão, V.M. Newly isolated phages preying on Pseudomonas syringae pv. garcae: In vitro and ex vivo inactivation studies in coffee plant leafs. Enzyme Microb. Technol. 2023, 171, 110325. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, L.M.R.; Almeida, I.M.G.; Patrício, F.R.A.; Beriam, L.O.S.; Maciel, K.W.; Braghini, M.T.; Guerreiro Filho, O. Mancha Aureolada do Cafeeiro Causada por Pseudomonas syringae pv. Garcae; Boletim Técnico IAC, Instituto Agronômico (IAC): Campinas, Brazil, 2013; 24p. Available online: https://www.iac.sp.gov.br/media/publicacoes/iacbt212.pdf (accessed on 5 June 2023).
- Rodrigues, L.M.R.; Queiroz-Voltan, R.B.; Guerreiro-Filho, O. Anatomical changes on coffee leaves infected by Pseudomonas syringae pv. garcae. Summa Phytopathol. 2015, 41, 256–261. [Google Scholar] [CrossRef]
- Rodrigues, L.M.R.; Sera, G.H.; Filho, O.G.; Beriam, L.O.S.; Almeida, I.M.G. First report of mixed infection by Pseudomonas syringae pathovars garcae and tabaci on coffee plantations. Plant Prot. 2017, 76, 543–549. [Google Scholar] [CrossRef]
- Belan, L.L.; Pozza, E.A.; Freitas, M.L.O.; Raimundi, M.K.; Souza, R.M.; Machado, J.C. Occurrence of Pseudomonas syringae pv. garcae in coffee seeds. Aust. J. Crop Sci. 2016, 10, 1015–1021. [Google Scholar] [CrossRef]
- Silva, F.J.; Vieira, B.S.; Siquieroli, A.C.S. Biological control of Pseudomonas syringae pv. garcae in coffee crop with Bacillus spp. isolates. Científica Jaboticabal 2019, 47, 364–370. [Google Scholar] [CrossRef]
- Souza, J.O.G.; Pozza, E.A.; Belam, L.L.; Freitas, M.L.O.; Oliveira, J.M. Initial Stages of infections process of Pseudomonas syringae pv garçae in coffee leaves. Coffee Sci. Lavras 2019, 14, 338–348. [Google Scholar] [CrossRef]
- Frampton, R.A.; Taylor, C.; Holguín Moreno, A.V.; Visnovsky, S.B.; Petty, N.K.; Pitman, A.R.; Fineran, P.C. Identification of Bacteriophages for Biocontrol of the Kiwifruit Canker Phytopathogen Pseudomonas syringae pv. actinidiae. Appl. Environ. Microbiol. 2014, 80, 2216–2228. [Google Scholar] [CrossRef]
- Jones, J.B.; Jackson, L.E.; Balogh, B.; Obradovic, A.; Iriarte, F.B.; Momol, M.T. Bacteriophages for Plant Disease Control. Annu. Rev. Phytopathol. 2007, 45, 245–262. [Google Scholar] [CrossRef]
- Harada, L.K.; Silva, E.C.; Campos, W.F.; Del Fiol, F.S.; Vila, M.M.D.C.; Dąbrowska, K.; Krylov, V.N.; Balcão, V.M. Biotechnological applications of bacteriophages: State of the art. Microbiol. Res. 2018, 212–213, 38–58. [Google Scholar] [CrossRef]
- Le, T.T.B. Bacteriophage: A Potential Treatment for Citrus Canker. Ph.D. Thesis, Texas A&M University, College Station, TX, USA, 2019. [Google Scholar]
- Svircev, A.; Roach, D.; Castle, A. Framing the future with bacteriophages in agriculture. Viruses 2018, 10, 218. [Google Scholar] [CrossRef]
- Żaczek, M.; Weber-Dabrowska, B.; Górski, A. Phages in the global fruit and vegetable industry. J. Appl. Microbiol. 2015, 118, 537–556. [Google Scholar] [CrossRef]
- Zaika, S.; Kharina, A.; Chaika, D. Looking for killers: Bacteriophages against phytopathogenic bacteria. Bull. Taras Shevchenko Natl. Univ. Kyiv 2013, 3, 86–89. [Google Scholar]
- Frampton, R.A.; Pitman, A.R.; Fineran, P.C. Advances in Bacteriophage-Mediated Control of Plant Pathogens. Int. J. Microbiol. 2012, 2012, 326452. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.; Bobrovnikov, D.G.; Ha, T. DNA mechanics and its biological impact. J. Mol. Biol. 2021, 433, 166861. [Google Scholar] [CrossRef]
- Tang, L. Sequencing DNA bendability. Nat. Methods 2021, 18, 121. [Google Scholar] [CrossRef] [PubMed]
- Dickerson, R.E. DNA bending: The prevalence of kinkiness and the virtues of normality. Nucleic Acids Res. 1998, 26, 1906–1926. [Google Scholar] [CrossRef] [PubMed]
- Balcão, V.M.; Basu, A.; Cieza, B.; Rossi, F.N.; Pereira, C.; Vila, M.M.D.C.; Setubal, J.C.; Ha, T.; da Silva, A.M. Pseudomonas-tailed lytic phages: Genome mechanical analysis and putative correlation with virion morphogenesis yield. Future Microbiol. 2022, 17, 1009–1026. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Bishop, A.R.; Rasmussen, K.Ø.; Alexandrov, B.S. The role of structural parameters in DNA cyclization. BMC Bioinform. 2016, 17, 68. [Google Scholar] [CrossRef]
- Vafabakhsh, R.; Ha, T. Extreme bendability of DNA less than 100 base pairs long revealed by single-molecule cyclization. Science 2012, 337, 1097. [Google Scholar] [CrossRef]
- Harada, L.K.; Silva, E.C.; Rossi, F.P.N.; Cieza, B.; Oliveira, T.J.; Pereira, C.; Tomazetto, G.; Silva, B.B.; Squina, F.M.; Vila, M.M.D.C.; et al. Characterization and in vitro testing of newly isolated lytic bacteriophages for biocontrol of Pseudomonas aeruginosa. Future Microbiol. 2022, 17, 111–141. [Google Scholar] [CrossRef]
- Basu, A.; Bobrovnikov, D.G.; Qureshi, Z.; Kayikcioglu, T.; Ngo, T.T.M.; Ranjan, A.; Eustermann, S.; Cieza, B.; Morgan, M.T.; Hejna, M.; et al. Measuring DNA mechanics on the genome scale. Nature 2021, 589, 462–467. [Google Scholar] [CrossRef] [PubMed]
- Balcão, V.M.; Moreli, F.C.; Silva, E.C.; Belline, B.G.; Martins, L.F.; Rossi, F.P.N.; Pereira, C.; Vila, M.M.D.C.; da Silva, A.M. Isolation and Molecular Characterization of a Novel Lytic Bacteriophage That Inactivates MDR Klebsiella pneumoniae Strains. Pharmaceutics 2022, 14, 1421. [Google Scholar] [CrossRef] [PubMed]
- Balcão, V.M.; Belline, B.G.; Silva, E.C.; Almeida, P.F.F.B.; Baldo, D.Â.; Amorim, L.R.P.; Oliveira Júnior, J.M.; Vila, M.M.D.C.; Del Fiol, F.S. Isolation and Molecular Characterization of Two Novel Lytic Bacteriophages for the Biocontrol of Escherichia coli in Uterine Infections: In Vitro and Ex Vivo Preliminary Studies in Veterinary Medicine. Pharmaceutics 2022, 14, 2344. [Google Scholar] [CrossRef]
- Kharina, A.V.; Zaika, S.A.; Yumyna, Y.M.; Zelena, P.P.; Kornienko, N.O.; Kosenko, Y.A.; Polischuk, V.P. Detection of Proteus mirabilis and Enterobacter cloacae in tomatto and pepper fruits and isolation of their bacteriophages. Наукoві Дoпoвіді Націoнальнoгo Університету Біoресурсів Прирoдoкoристування України 2015, 13, 80–93. [Google Scholar]
- Ewing, B.; Hillier, L.; Wendl, M.C.; Green, P. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 1998, 8, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [PubMed]
- Kokot, M.; Dlugosz, M.; Deorowicz, S. KMC 3: Counting and manipulating k-mer statistics. Bioinformatics 2017, 33, 2759–2761. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Song, L.; Florea, L.; Langmead, B. Lighter: Fast and memory-efficient sequencing error correction without counting. Genome Biol. 2014, 15, 509. [Google Scholar] [CrossRef]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [PubMed]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Rincon, N.; Wood, D.E.; Breitwieser, F.P.; Pockrandt, C.; Langmead, B.; Salzberg, S.L.; Steinegger, M. Metagenome analysis using the Kraken software suite. Nat. Protoc. 2022, 17, 2815–2839. [Google Scholar] [CrossRef]
- Nishimura, Y.; Watai, H.; Honda, T.; Mihara, T.; Omae, K.; Roux, S.; Blanc-Mathieu, R.; Yamamoto, K.; Hingamp, P.; Sako, Y.; et al. Environmental Viral Genomes Shed New Light on Virus-Host Interactions in the Ocean. mSphere 2017, 2, e00359-16. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Bouras, G.; Nepal, R.; Houtak, G.; Psaltis, A.J.; Wormald, P.J.; Vreugde, S. Pharokka: A fast scalable bacteriophage annotation tool. Bioinformatics 2023, 39, btac776. [Google Scholar] [CrossRef]
- Chan, P.P.; Lin, B.Y.; Mak, A.J.; Lowe, T.M. tRNAscan-SE 2.0: Improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 2021, 49, 9077–9096. [Google Scholar] [CrossRef]
- Laslett, D.; Canback, B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004, 32, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Bland, C.; Ramsey, T.L.; Sabree, F.; Lowe, M.; Brown, K.; Kyrpides, N.C.; Hugenholtz, P. CRISPR Recognition Tool (CRT): A tool for automatic detection of clustered regularly interspaced palindromic repeats. BMC Bioinform. 2007, 8, 209. [Google Scholar] [CrossRef] [PubMed]
- McNair, K.; Zhou, C.; Dinsdale, E.A.; Souza, B.; Edwards, R.A. PHANOTATE: A novel approach to gene identification in phage genomes. Bioinformatics 2019, 35, 4537–4542. [Google Scholar] [CrossRef]
- Mirdita, M.; Steinegger, M.; Breitwieser, F.; Söding, J.; Levy Karin, E. Fast and sensitive taxonomic assignment to metagenomic contigs. Bioinformatics 2021, 37, 3029–3031. [Google Scholar] [CrossRef] [PubMed]
- Terzian, P.; Ndela, E.O.; Galiez, C.; Lossouarn, J.; Bucio, R.E.P.; Mom, R.; Toussaint, A.; Petit, M.-A.; Enault, F. PHROG: Families of prokaryotic virus proteins clustered using remote homology. NAR Genom. Bioinform. 2021, 3, lqab067. [Google Scholar] [CrossRef]
- Alcock, B.P.; Huynh, W.; Chalil, R.; Smith, K.W.; Raphenya, A.R.; Wlodarski, M.A.; Edalatmand, A.; Petkau, A.; Syed, S.A.; Tsang, K.K.; et al. CARD 2023: Expanded curation, support for machine learning, and resistome prediction at the Comprehensive Antibiotic Resistance Database. Nucleic Acids Res. 2023, 51, D690–D699. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zheng, D.; Zhou, S.; Chen, L.; Yang, J. VFDB 2022: A general classification scheme for bacterial virulence factors. Nucleic Acids Res. 2022, 50, D912–D917. [Google Scholar] [CrossRef] [PubMed]
- Schwengers, O.; Hain, T.; Chakraborty, T.; Goesmann, A. ReferenceSeeker: Rapid determination of appropriate reference genomes. J. Open Source Softw. 2020, 5, 1994. [Google Scholar] [CrossRef]
- Ondov, B.D.; Treangen, T.J.; Melsted, P.; Mallonee, A.B.; Bergman, N.H.; Koren, S.; Phillippy, A.M. Mash: Fast genome and metagenome distance estimation using MinHash. Genome Biol. 2016, 17, 132. [Google Scholar] [CrossRef]
- Marçais, G.; Delcher, A.L.; Phillippy, A.M.; Coston, R.; Salzberg, S.L.; Zimin, A. MUMmer4: A fast and versatile genome alignment system. PLoS Comput. Biol. 2018, 14, e1005944. [Google Scholar] [CrossRef]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef] [PubMed]
- Katz, L.S.; Griswold, T.; Morrison, S.S.; Caravas, J.A.; Zhang, S.; den Bakker, H.C.; Deng, X.; Carleton, H.A. Mashtree: A rapid comparison of whole genome sequence files. J. Open Source Softw. 2019, 4, 1762. [Google Scholar] [CrossRef]
- Howe, K.; Bateman, A.; Durbin, R. QuickTree: Building huge Neighbour-Joining trees of protein sequences. Bioinformatics 2002, 18, 1546–1547. [Google Scholar] [CrossRef] [PubMed]
- Bin Jang, H.; Bolduc, B.; Zablocki, O.; Kuhn, J.H.; Roux, S.; Adriaenssens, E.M.; Brister, J.R.; Kropinski, A.M.; Krupovic, M.; Lavigne, R.; et al. Taxonomic assignment of uncultivated prokaryotic virus genomes is enabled by gene-sharing networks. Nat. Biotechnol. 2019, 37, 632–639. [Google Scholar] [CrossRef] [PubMed]
- Cook, R.; Brown, N.; Redgwell, T.; Rihtman, B.; Barnes, M.; Clokie, M.; Stekel, D.J.; Hobman, J.; Jones, M.A.; Millard, A. INfrastructure for a PHAge REference Database: Identification of Large-Scale Biases in the Current Collection of Cultured Phage Genomes. Phage 2021, 2, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree: Computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 2009, 26, 1641–1650. [Google Scholar] [CrossRef] [PubMed]
- Whelan, S.; Goldman, N. A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol. Biol. Evol. 2001, 18, 691–699. [Google Scholar] [CrossRef]
- Gascuel, O. On the optimization principle in phylogenetic analysis and the minimum-evolution criterion. Mol. Biol. Evol. 2000, 17, 401–405. [Google Scholar] [CrossRef]
- Zhou, T.; Yang, L.; Lu, Y.; Dror, I.; Machado, A.C.D.; Ghane, T.; Di Felice, R.; Rohs, R. DNAshape: A method for the high-throughput prediction of DNA structural features on a genomic scale. Nucleic Acids Res. 2013, 41, W56–W62. [Google Scholar] [CrossRef]
- Basu, A.; Bobrovnikov, D.G.; Cieza, B.; Arcon, J.P.; Qureshi, Z.; Orozco, M.; Ha, T. Deciphering the mechanical code of the genome and epigenome. Nat. Struct. Mol. Biol. 2022, 29, 1178–1187. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Basu, A.; Ha, T.; Bialek, W. Searching for sequence features that control DNA flexibility. arXiv 2020, arXiv:2012.06127v1. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Turner, D.; Shkoporov, A.N.; Lood, C.; Millard, A.D.; Dutilh, B.E.; Alfenas-Zerbini, P.; van Zyl, L.J.; Aziz, R.K.; Oksanen, H.M.; Poranen, M.M.; et al. Abolishment of morphology-based taxa and change to binomial species names: 2022 taxonomy update of the ICTV bacterial viruses subcommittee. Arch. Virol. 2023, 168, 74. [Google Scholar] [CrossRef]
- Nishimura, Y.; Yoshida, T.; Kuronishi, M.; Uehara, H.; Ogata, H.; Goto, S. ViPTree: The viral proteomic tree server. Bioinformatics 2017, 33, 2379–2380. [Google Scholar] [CrossRef] [PubMed]
- Xuan, G.; Kong, J.; Wang, Y.; Lin, H.; Wang, J. Characterization of the newly isolated Pseudomonas phage vB_Pae_LC3I3. Virus Res. 2023, 323, 198978. [Google Scholar] [CrossRef] [PubMed]
- UniProt Consortium UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [CrossRef]
- Schoch, C.L.; Ciufo, S.; Domrachev, M.; Hotton, C.L.; Kannan, S.; Khovanskaya, R.; Leipe, D.; Mcveigh, R.; O’Neill, K.; Robbertse, B.; et al. NCBI Taxonomy: A comprehensive update on curation, resources and tools. Database 2020, 2020, baaa062. [Google Scholar] [CrossRef]
- Hyman, P. Phages for Phage Therapy: Isolation, Characterization, and Host Range Breadth. Pharmaceuticals 2019, 12, 35. [Google Scholar] [CrossRef]
- Kakasis, A.; Panitsa, G. Bacteriophage therapy as an alternative treatment for human infections. A comprehensive review. Int. J. Antimicrob. Agents 2019, 53, 16–21. [Google Scholar] [CrossRef]
- Wintachai, P.; Surachat, K.; Chaimaha, G.; Septama, A.W.; Smith, D.R. Isolation and Characterization of a Phapecoctavirus Infecting Multidrug-Resistant Acinetobacter baumannii in A549 Alveolar Epithelial Cells. Viruses 2022, 14, 2561. [Google Scholar] [CrossRef] [PubMed]
- Khalifeh, A.; Kraberger, S.; Dziewulska, D.; Stenzel, T.; Varsani, A. Complete Genome Sequence of a Phapecoctavirus Isolated from a Pigeon Cloacal Swab Sample. Microbiol. Resour. Announc. 2021, 10, e01471-20. [Google Scholar] [CrossRef] [PubMed]
- Vasquez, I.; Retamales, J.; Parra, B.; Machimbirike, V.; Robeson, J.; Santander, J. Comparative genomics of a polyvalent Escherichia-Salmonella phage fp01 and in silico analysis of its receptor binding protein and conserved Enterobacteriaceae phage receptor. Viruses 2023, 15, 379. [Google Scholar] [CrossRef] [PubMed]
- Abdelhadi, I.M.A.; Sofy, A.R.; Hmed, A.A.; Refaey, E.E.; Soweha, H.E.; Abbas, M.A. Discovery of polyvalent myovirus (vB_STM-2) phage as a natural antimicrobial system to lysis and biofilm removal of Salmonella typhimurium isolates from various food sources. Sustainability 2021, 13, 11602. [Google Scholar] [CrossRef]
- Kim, S.-H.; Adeyemi, D.E.; Park, M.-K. Characterization of a new and efficient polyvalent phage infecting E. coli O157:H7, Salmonella spp., and Shigella sonnei. Microorganisms 2021, 9, 2105. [Google Scholar] [CrossRef] [PubMed]
- Sui, B.; Han, L.; Ren, H.; Liu, W.; Zhang, C. A novel polyvalent bacteriophage vB_EcoM_swi3 infects pathogenic Escherichia coli and Salmonella enteritidis. Front. Microbiol. 2021, 12, 649673. [Google Scholar] [CrossRef]
- Iyer, L.M.; Burroughs, A.M.; Anand, S.; de Souza, R.F.; Aravind, L. Polyvalent proteins, a pervasive theme in the intergenomic biological conflicts of bacteriophages and conjugative elements. J. Bacteriol. 2017, 199, e00245-17. [Google Scholar] [CrossRef]
- Parra, B.; Robeson, J. Selection of polyvalent bacteriophages infecting Salmonella enterica serovar Choleraesuis. Electron. J. Biotechnol. 2016, 21, 72–76. [Google Scholar] [CrossRef]
- Yu, P.; Mathieu, J.; Li, M.; Dai, Z.; Alvarez, P.J.J. Isolation of polyvalent bacteriophages by sequential multiple-host approaches. Appl. Environ. Microbiol. 2016, 82, 808–815. [Google Scholar] [CrossRef]
- Zhou, T.; Shen, N.; Yang, L.; Abe, N.; Horton, J.; Mann, R.S.; Bussemaker, H.J.; Gordân, R.; Rohs, R. Quantitative modeling of transcription factor binding specificities using DNA shape. Proc. Natl. Acad. Sci. USA 2015, 112, 4654–4659. [Google Scholar] [CrossRef]
- Ussery, D.W. DNA Structure: A-, B- and Z-DNA Helix Families. Encycl. Life Sci. 2002, 1, e003122. [Google Scholar] [CrossRef]
- Rao, S.; Chiu, T.P.; Kribelbauer, J.F.; Mann, R.S.; Bussemaker, H.J.; Rohs, R. Systematic prediction of DNA shape changes due to CpG methylation explains epigenetic effects on protein-DNA binding. Epigenetics Chromatin 2018, 11, 6. [Google Scholar] [CrossRef] [PubMed]
- Rohs, R.; West, S.M.; Sosinsky, A.; Liu, P.; Mann, R.S.; Honig, B. The role of DNA shape in protein-DNA recognition. Nature 2009, 461, 1248–1253. [Google Scholar] [CrossRef]
- Parker, A.V.; Mann, D.; Tzokov, S.B.; Hwang, L.C.; Bergeron, J.R.C. The structure of the bacterial DNA segregation ATPase filament reveals the conformational plasticity of ParA upon DNA binding. Nat. Commun. 2021, 12, 5166. [Google Scholar] [CrossRef]
- Bharati, A.P.; Kashif, M.; Chaturvedi, S.K.; Khan, R.H.; Ahmad, A. An insight into structural plasticity and conformational transitions of transcriptional co-activator Sus1. PLoS ONE 2020, 15, e0229216. [Google Scholar] [CrossRef]
- Fratini, A.V.; Kopka, M.L.; Drew, H.R.; Dickerson, R.E. Reversible bending and helix geometry in a B-DNA dodecamer: CGCGAATTBrCGCG. J. Biol. Chem. 1982, 257, 14686–14707. [Google Scholar] [PubMed]
- Liebl, K.; Drsata, T.; Lankas, F.; Lipfert, J.; Zacharias, M. Explaining the striking difference in twist-stretch coupling between DNA and RNA: A comparative molecular dynamics analysis. Nucleic Acids Res. 2015, 43, 10143–10156. [Google Scholar] [CrossRef]
- El Hassan, M.A.; Calladine, C.R. Propeller-twisting of base-pairs and the conformational mobility of dinucleotide steps in DNA. J. Mol. Biol. 1996, 259, 95–103. [Google Scholar] [CrossRef]
- Yella, V.R.; Bhimsaria, D.; Ghoshdastidar, D.; Rodríguez-Martínez, J.A.; Ansari, A.Z.; Bansal, M. Flexibility and structure of flanking DNA impact transcription factor affinity for its core motif. Nucleic Acids Res. 2018, 46, 11883–11897. [Google Scholar] [CrossRef]
- Wang, D.D.; Yan, H. The relationship between periodic dinucleotides and the nucleosomal DNA deformation revealed by normal mode analysis. Phys. Biol. 2011, 8, 066004. [Google Scholar] [CrossRef]
- Gu, H.; Fan, R.L.Y.; Wang, D.; Poon, L.L.M. Dinucleotide evolutionary dynamics in influenza A virus. Virus Evol. 2019, 5, vez038. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.-F.; Virk, N.; Wang, H.-Z. Chapter 19—Impact of the host on plant virus evolution. In Plant Virus–Host Interaction; Gaur, R.K., Hohn, T., Sharma, P., Eds.; Academic Press: Cambridge, MA, USA, 2014; pp. 359–371. ISBN 9780124115842. [Google Scholar]
- Karlin, S.; Mrázek, J. Compositional differences within and between eukaryotic genomes. Proc. Natl. Acad. Sci. USA 1997, 94, 10227–10232. [Google Scholar] [CrossRef] [PubMed]
- Di Giallonardo, F.; Schlub, T.E.; Shi, M.; Holmes, E.C. Dinucleotide Composition in Animal RNA Viruses Is Shaped More by Virus Family than by Host Species. J. Virol. 2017, 91, e02381-16. [Google Scholar] [CrossRef]
- Wu, Q.; Zhou, W.; Wang, J.; Yan, H. Correlation between the flexibility and periodic dinucleotide patterns in yeast nucleosomal DNA sequences. J. Theor. Biol. 2011, 284, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Okonogi, T.M.; Alley, S.C.; Reese, A.W.; Hopkins, P.B.; Robinson, B.H. Sequence-dependent dynamics of duplex DNA: The applicability of a dinucleotide model. Biophys. J. 2002, 83, 3446–3459. [Google Scholar] [CrossRef]
- Mrázek, J. Comparative analysis of sequence periodicity among prokaryotic genomes points to differences in nucleoid structure and a relationship to gene expression. J. Bacteriol. 2010, 192, 3763–3772. [Google Scholar] [CrossRef]
- Johnson, S.; Chen, Y.J.; Phillips, R. Poly(dA:dT)-rich DNAs are highly flexible in the context of DNA looping. PLoS ONE 2013, 8, e75799. [Google Scholar] [CrossRef]
- Packer, M.J.; Dauncey, M.P.; Hunter, C.A. Sequence-dependent DNA structure: Dinucleotide conformational maps. J. Mol. Biol. 2000, 295, 71–83. [Google Scholar] [CrossRef]
- Back, G.; Walther, D. Predictions of DNA mechanical properties at a genomic scale reveal potentially new functional roles of DNA flexibility. NAR Genom. Bioinform. 2023, 5, lqad097. [Google Scholar] [CrossRef]
- Lyubchenko, Y.L.; Shlyakhtenko, L.S.; Appella, E.; Harrington, R.E. CA runs increase DNA flexibility in the complex of lambda Cro protein with the OR3 site. Biochemistry 1993, 32, 4121–4127. [Google Scholar] [CrossRef]
- Burge, C.; Campbell, A.M.; Karlin, S. Over- and under-representation of short oligonucleotides in DNA sequences. Proc. Natl. Acad. Sci. USA 1992, 89, 1358–1362. [Google Scholar] [CrossRef] [PubMed]
- Travers, A.A. The structural basis of DNA flexibility. Philos. Trans. A Math. Phys. Eng. Sci. 2004, 362, 1423–1438. [Google Scholar] [CrossRef] [PubMed]
- El Hassan, M.A.; Calladine, C.R. Conformational characteristics of DNA: Empirical classifications and a hypothesis for the conformational behavior of dinucleotide steps. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 1997, 355, 43–100. [Google Scholar] [CrossRef]
- Langowski, J.; Fujimoto, B.S.; Wemmer, D.E.; Benight, A.S.; Drobny, G.; Shibata, J.H.; Schurr, J.M. Deformational dynamics and nmr relaxation of supercoiled DNAs. Biopolymers 1985, 24, 1023–1056. [Google Scholar] [CrossRef] [PubMed]
- Shishkin, O.V.; Gorb, L.; Leszczynski, J. Conformational flexibility of pyrimidine ring in adenine and related compounds. Chem. Phys. Lett. 2000, 330, 603–611. [Google Scholar] [CrossRef]
- Geggier, S.; Vologodskii, A. Sequence dependence of DNA bending rigidity. Proc. Natl. Acad. Sci. USA 2010, 107, 15421–15426. [Google Scholar] [CrossRef]
- Protozanova, E.; Yakovchuk, P.; Frank-Kamenetskii, M.D. Stacked-unstacked equilibrium at the nick site of DNA. J. Mol. Biol. 2004, 342, 775–785. [Google Scholar] [CrossRef]
- Crothers, D.M.; Haran, T.E.; Nadeau, J.G. Intrinsically bent DNA. J. Biol. Chem. 1990, 265, 7093–7096. [Google Scholar] [CrossRef]
- Hu, E.Z.; Lan, X.R.; Liu, Z.L.; Gao, J.; Niu, D.K. A positive correlation between GC content and growth temperature in prokaryotes. BMC Genom. 2022, 23, 110. [Google Scholar] [CrossRef]
- Shishkin, O.V.; Šponer, J.; Hobza, P. Intramolecular flexibility of DNA bases in adenine–thymine and guanine–cytosine Watson–Crick base pairs. J. Mol. Struct. 1999, 477, 15–21. [Google Scholar] [CrossRef]
- Jeong, J.; Kim, H.D. Determinants of cyclization-decyclization kinetics of short DNA with sticky ends. Nucleic Acids Res. 2020, 48, 5147–5156. [Google Scholar] [CrossRef] [PubMed]
- Du, Q.; Vologodskaia, M.; Kuhn, H.; Frank-Kamenetskii, M.; Vologodskii, A. Gapped DNA and cyclization of short DNA fragments. Biophys. J. 2005, 88, 4137–4145. [Google Scholar] [CrossRef] [PubMed]
- Shore, D.; Baldwin, R.L. Energetics of DNA twisting. I. Relation between twist and cyclization probability. J. Mol. Biol. 1983, 170, 957–981. [Google Scholar] [CrossRef] [PubMed]
- Podtelezhnikov, A.A.; Mao, C.; Seeman, N.C.; Vologodskii, A. Multimerization-cyclization of DNA fragments as a method of conformational analysis. Biophys. J. 2000, 79, 2692–2704. [Google Scholar] [CrossRef]
- Rao, V.B.; Feiss, M. Mechanisms of DNA Packaging by Large Double-Stranded DNA Viruses. Annu. Rev. Virol. 2015, 2, 351–378. [Google Scholar] [CrossRef]
- Smug, B.J.; Szczepaniak, K.; Rocha, E.P.C.; Dunin-Horkawicz, S.; Mostowy, R.J. Ongoing shuffling of protein fragments diversifies core viral functions linked to interactions with bacterial hosts. Nat. Commun. 2023, 14, 7460. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structural Feature | Phage Virion Dimensions | |
|---|---|---|
| Phage PsgM02F | Phage PsgM04F | |
| Capsid length (nm) | 101.3 ± 4.0 | 77.5 ± 3.5 |
| Capsid width (nm) | 81.5 ± 2.8 | 73.0 ± 2.2 |
| Tail length (nm) | 108.7 ± 0.8 | 102.5 ± 4.3 |
| Tail thickness (nm) | 21.9 ± 3.0 | 15.6 ± 3.8 |
| Feature | Phage PsgM02F | Phage PsgM04F |
|---|---|---|
| NCBI/Genbank accession number | OR584013 | OR584014 |
| Genome size | 165,282 bp | 151,205 bp |
| Number of PE reads mapping in the final assembly | 47,224,300 (91.64%) | 25,251,477 (99.48%) |
| Average sequencing coverage (calculated as (length of reads × number of reads mapping)/genome size) | 171,636× | 33,443× |
| GC content | 35.37% | 42.30% |
| tRNA genes | 11 | 11 |
| Protein-coding genes (CDS) predicted | 278 | 324 |
| • With function assigned | 136 (48.9%) | 73 (22.5%) |
| • Hypothetical/unknown function | 142 (51.1%) | 251 (77.5%) |
| Similar phage genome sequences NCBI/RefSeq accession number of similar phage | Escherichia phage HP3/Shigella phage Sf21 GCF_002619885.1/GCF_002955385.1 | Escherichia phage phiWec190 LC739539.1 |
| Morphotype | Myovirus | Myovirus |
| Family/Genus | Straboviridae/Tequatrovirus | Stephanstirmvirinae/Phapecoctavirus |
| Source of Variation | SS | df | MS | F-Ratio | p-Value | F-Critical |
|---|---|---|---|---|---|---|
| Between Groups | 17.288 | 1.000 | 17.288 | 428.585 | 9.105 × 10−95 | 3.842 |
| Within Groups | 1823.138 | 45,197 | 0.040 | |||
| Total | 1840.426 | 45,198 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silva, E.C.; Quinde, C.A.; Cieza, B.; Basu, A.; Vila, M.M.D.C.; Balcão, V.M. Molecular Characterization and Genome Mechanical Features of Two Newly Isolated Polyvalent Bacteriophages Infecting Pseudomonas syringae pv. garcae. Genes 2024, 15, 113. https://doi.org/10.3390/genes15010113
Silva EC, Quinde CA, Cieza B, Basu A, Vila MMDC, Balcão VM. Molecular Characterization and Genome Mechanical Features of Two Newly Isolated Polyvalent Bacteriophages Infecting Pseudomonas syringae pv. garcae. Genes. 2024; 15(1):113. https://doi.org/10.3390/genes15010113
Chicago/Turabian StyleSilva, Erica C., Carlos A. Quinde, Basilio Cieza, Aakash Basu, Marta M. D. C. Vila, and Victor M. Balcão. 2024. "Molecular Characterization and Genome Mechanical Features of Two Newly Isolated Polyvalent Bacteriophages Infecting Pseudomonas syringae pv. garcae" Genes 15, no. 1: 113. https://doi.org/10.3390/genes15010113