
Conceptualizing Epigenetics and the Environmental Landscape of Autism Spectrum Disorders
 , ,
, ,
Abstract
:1. Clinical Vignette Case
2. Introduction
3. Epigenetics: An Overview
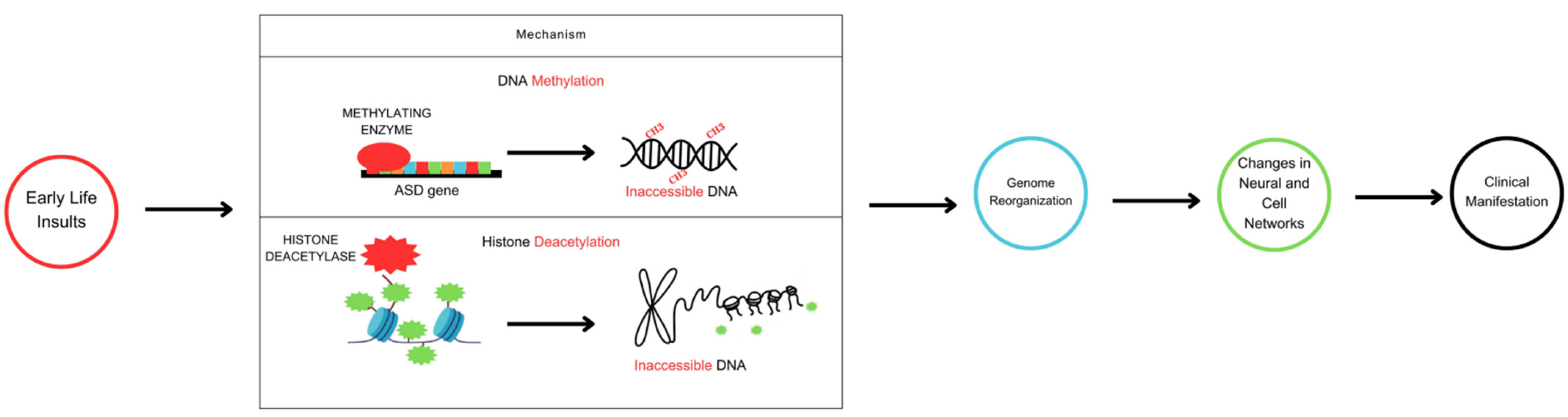
4. DNA Methylation and Histone Deacetylation in ASD
5. Homeodomain-Based Strategies for the Identification of Genes Associated with ASD
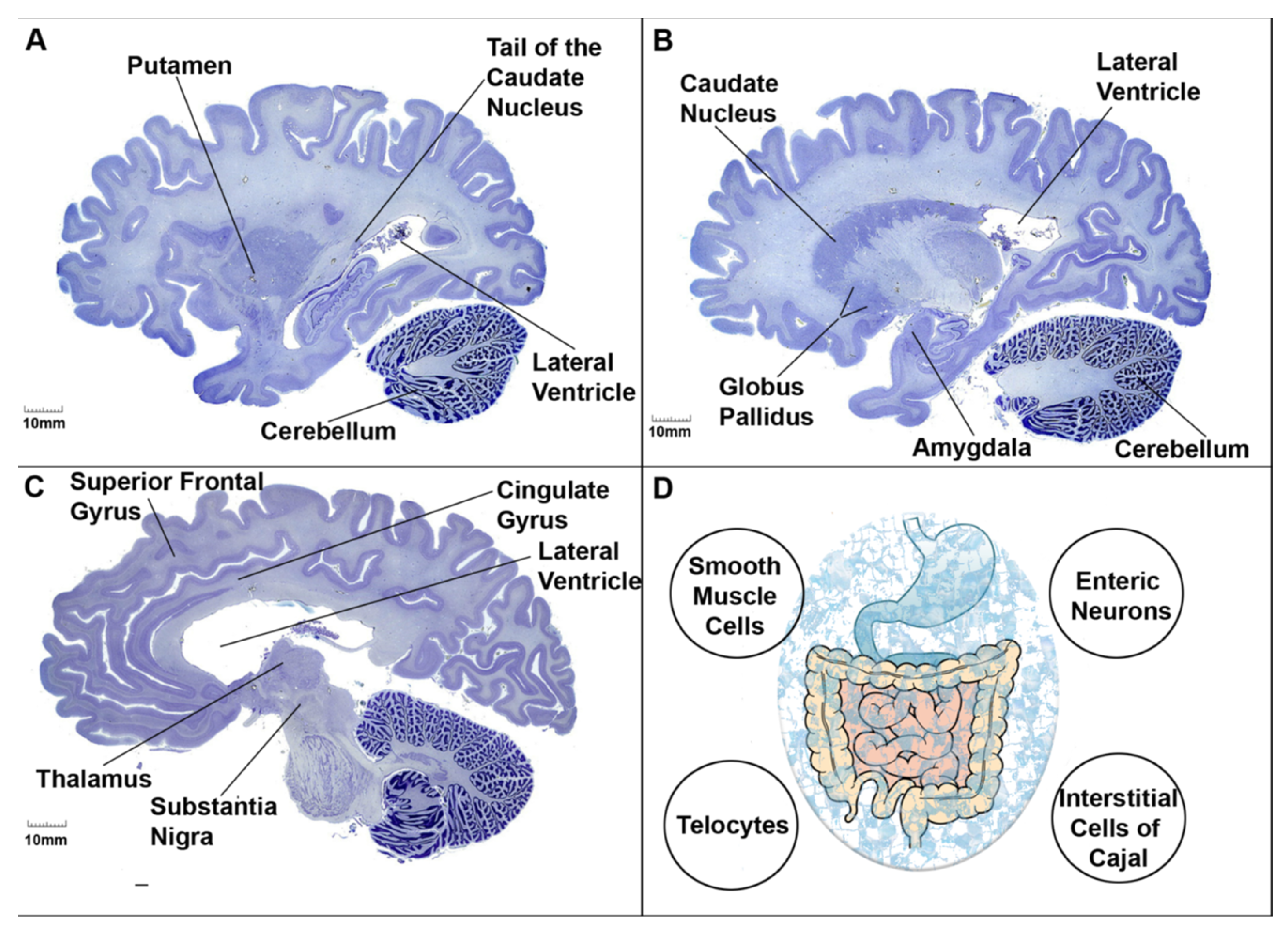
6. Clinical Case Presentation of ASD
7. Genomic Imprinting in ASD
8. Sex-Dependent Differences in ASD
9. Assisted Reproductive Technology and ASD Risks
10. Evolution of Autistic Spectrum Disorders
11. Epigenetics and Autoimmunity in ASD
12. Outlook on Epigenetic Diagnostics and Treatment of ASD
13. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lecavalier, L.; McCracken, C.E.; Aman, M.G.; McDougle, C.J.; McCracken, J.T.; Tierney, E.; Smith, T.; Johnson, C.; King, B.; Handen, B.; et al. An exploration of concomitant psychiatric disorders in children with autism spectrum disorder. Compr. Psychiatry 2019, 88, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Kristan, W.B., Jr. Early evolution of neurons. Curr. Biol. 2016, 26, R949–R954. [Google Scholar] [CrossRef] [PubMed]
- Dimaline, R.; Dockray, G.J. Evolution of the gastrointestinal endocrine system (with special reference to gastrin and CCK). Baillieres Clin. Endocrinol. Metab. 1994, 8, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Harvey, Z.H.; Chen, Y.; Jarosz, D.F. Protein-Based Inheritance: Epigenetics beyond the Chromosome. Mol. Cell 2018, 69, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.Y.; Aromolaran, K.A.; Zukin, R.S. The emerging field of epigenetics in neurodegeneration and neuroprotection. Nat. Rev. Neurosci. 2017, 18, 347–361, Erratum in: Nat. Rev. Neurosci. 2018, 19, 771. [Google Scholar] [CrossRef]
- Jeffries, M.A. The Development of Epigenetics in the Study of Disease Pathogenesis. Adv. Exp. Med. Biol. 2020, 1253, 57–94. [Google Scholar] [CrossRef]
- Tseng, C.J.; McDougle, C.J.; Hooker, J.M.; Zürcher, N.R. Epigenetics of Autism Spectrum Disorder: Histone Deacetylases. Biol. Psychiatry 2022, 91, 922–933. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Liang, F.; Meng, G.; Nie, Z.; Zhou, R.; Cheng, W.; Wu, X.; Feng, Y.; Wang, Y. Redox/methylation mediated abnormal DNA methylation as regulators of ambient fine particulate matter-induced neurodevelopment related impairment in human neuronal cells. Sci. Rep. 2016, 6, 33402. [Google Scholar] [CrossRef]
- Nardone, S.; Elliott, E. The Interaction between the Immune System and Epigenetics in the Etiology of Autism Spectrum Disorders. Front. Neurosci. 2016, 10, 329. [Google Scholar] [PubMed]
- De Rubeis, S.; Siper, P.M.; Durkin, A.; Weissman, J.; Muratet, F.; Halpern, D.; Trelles, M.D.P.; Frank, Y.; Lozano, R.; Wang, A.T.; et al. Delineation of the genetic and clinical spectrum of Phelan-McDermid syndrome caused by SHANK3 point mutations. Mol. Autism 2018, 9, 31. [Google Scholar] [CrossRef]
- Kimura, R.; Nakata, M.; Funabiki, Y.; Suzuki, S.; Awaya, T.; Murai, T.; Hagiwara, M. An epigenetic biomarker for adult high-functioning autism spectrum disorder. Sci. Rep. 2019, 9, 13662. [Google Scholar] [CrossRef] [PubMed]
- Masini, E.; Loi, E.; Vega-Benedetti, A.F.; Carta, M.; Doneddu, G.; Fadda, R.; Zavattari, P. An Overview of the Main Genetic, Epigenetic and Environmental Factors Involved in Autism Spectrum Disorder Focusing on Synaptic Activity. Int. J. Mol. Sci. 2020, 21, 8290. [Google Scholar] [CrossRef]
- Stathopoulos, S.; Gaujoux, R.; Lindeque, Z.; Mahony, C.; Van Der Colff, R.; Van Der Westhuizen, F.; O’Ryan, C. DNA Methylation Associated with Mitochondrial Dysfunction in a South African Autism Spectrum Disorder Cohort. Autism Res. 2020, 13, 1079–1093. [Google Scholar] [CrossRef] [PubMed]
- Bekiesinska-Figatowska, M.; Mierzewska, H.; Jurkiewicz, E. Basal ganglia lesions in children and adults. Eur. J. Radiol. 2013, 82, 837–849. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.M.; Torregrossa, M.M. Valence encoding in the amygdala influences motivated behavior. Behav. Brain. Res. 2021, 411, 113370. [Google Scholar] [CrossRef]
- Mouat, J.S.; LaSalle, J.M. The Promise of DNA Methylation in Understanding Multigenerational Factors in Autism Spectrum Disorders. Front. Genet. 2022, 13, 831221. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Poschmann, J.; Cruz-Herrera Del Rosario, R.; Parikshak, N.N.; Hajan, H.S.; Kumar, V.; Ramasamy, R.; Belgard, T.G.; Elanggovan, B.; Wong, C.C.Y.; et al. Histone Acetylome-wide Association Study of Autism Spectrum Disorder. Cell 2016, 167, 1385–1397.e11. [Google Scholar] [CrossRef] [PubMed]
- Waye, M.M.Y.; Cheng, H.Y. Genetics and epigenetics of autism: A review. Psychiatry Clin. Neurosci. 2018, 72, 228–244. [Google Scholar] [CrossRef]
- GeneCards. Autism Genes. Available online: https://www.genecards.org/Search/Keyword?queryString=autism%20genes (accessed on 30 June 2023).
- GeneCards. Homeodomain Genes. Available online: https://www.genecards.org/Search/Keyword?queryString=homeodomain%20genes (accessed on 30 June 2023).
- GeneCards. Brain Development Genes. Available online: https://www.genecards.org/Search/Keyword?queryString=brain%20development%20genes (accessed on 30 June 2023).
- UCSC Genome Browser. ZEB1 Interacting Genes. Available online: https://genome.ucsc.edu/cgi-bin/hgGeneGraph?gene=ZEB1&1=OK&supportLevel=text&geneCount=25 (accessed on 30 June 2023).
- UCSC Genome Browser. PBX1 Interacting Genes. Available online: https://genome.ucsc.edu/cgi-bin/hgGeneGraph?gene=PBX1&1=OK&supportLevel=text&geneCount=25&1=OK&geneCount=25 (accessed on 30 June 2023).
- UCSC Genome Browser. NKX2-1 Interacting Genes. Available online: https://genome.ucsc.edu/cgi-bin/hgGeneGraph?gene=NKX2-1&1=OK&supportLevel=text&geneCount=25&1=OK&geneCount=25 (accessed on 30 June 2023).
- UCSC Genome Browser. ZEB2 Interacting Genes. Available online: https://genome.ucsc.edu/cgi-bin/hgGeneGraph?gene=ZEB2&1=OK&supportLevel=text&geneCount=25&1=OK&geneCount=25 (accessed on 30 June 2023).
- UCSC Genome Browser. OTX2 Interacting Genes. Available online: https://genome.ucsc.edu/cgi-bin/hgGeneGraph?gene=OTX2&1=OK&supportLevel=text&geneCount=25&1=OK&geneCount=25 (accessed on 30 June 2023).
- UCSC Genome Browser. NKX2-5 Interacting Genes. Available online: https://genome.ucsc.edu/cgi-bin/hgGeneGraph?gene=NKX2-5&1=OK&supportLevel=text&geneCount=25&1=OK&geneCount=25 (accessed on 30 June 2023).
- UCSC Genome Browser. SATB2 Interacting Genes. Available online: https://genome.ucsc.edu/cgi-bin/hgGeneGraph?gene=SATB2&1=OK&supportLevel=text&geneCount=25&1=OK&geneCount=25 (accessed on 30 June 2023).
- UCSC Genome Browser. CUX1 Interacting Genes. Available online: https://genome.ucsc.edu/cgi-bin/hgGeneGraph?gene=CUX1&1=OK&supportLevel=text&geneCount=25&1=OK&geneCount=25 (accessed on 30 June 2023).
- UCSC Genome Browser. EMX2 Interacting Genes. Available online: https://genome.ucsc.edu/cgi-bin/hgGeneGraph?gene=EMX2&1=OK&supportLevel=text&geneCount=25&1=OK&geneCount=25 (accessed on 30 June 2023).
- UCSC Genome Browser. ARX Interacting Genes. Available online: https://genome.ucsc.edu/cgi-bin/hgGeneGraph?gene=ARX&1=OK&supportLevel=text&geneCount=25&1=OK&geneCount=25 (accessed on 30 June 2023).
- UCSC Genome Browser. SIX3 Interacting Genes. Available online: https://genome.ucsc.edu/cgi-bin/hgGeneGraph?gene=SIX3&1=OK&supportLevel=text&geneCount=25&1=OK&geneCount=25 (accessed on 30 June 2023).
- UCSC Genome Browser. ADNP Interacting Genes. Available online: https://genome.ucsc.edu/cgi-bin/hgGeneGraph?gene=ADNP&1=OK&supportLevel=text&geneCount=25&1=OK&geneCount=25 (accessed on 30 June 2023).
- UCSC Genome Browser. DLX5 Interacting Genes. Available online: https://genome.ucsc.edu/cgi-bin/hgGeneGraph?gene=DLX5&1=OK&supportLevel=text&geneCount=25&1=OK&geneCount=25 (accessed on 30 June 2023).
- UCSC Genome Browser. LMX1B Interacting Genes. Available online: https://genome.ucsc.edu/cgi-bin/hgGeneGraph?gene=LMX1B&1=OK&supportLevel=text&geneCount=25&1=OK&geneCount=25 (accessed on 30 June 2023).
- Kolesnikoff, N.; Attema, J.L.; Suraya Roslan, S.; Bert, A.G.; Schwarz, Q.P.; Gregory PA and Goodall, G.J. Specificity protein 1 (Sp1) maintains basal epithelial expression of the miR-200 family: Implications for epithelial-mesenchymal transition. J. Biol. Chem. 2014, 289, 11194–11205. [Google Scholar] [CrossRef]
- Li, C.; Ling, X.; Yuan, B.; Minoo, P. A novel DNA element mediates transcription of Nkx2.1 by Sp1 and Sp3 in pulmonary epithelial cells. Comp. Study Biochim. Biophys. Acta 2000, 1490, 213–224. [Google Scholar] [CrossRef]
- Lu, Q.; Kamps, M.P. Selective repression of transcriptional activators by Pbx1 does not require the homeodomain. Proc. Natl. Acad. Sci. USA 1996, 93, 470–474. [Google Scholar] [CrossRef] [PubMed]
- Nam, E.; Lee, Y.; Zhao, X.; Park, Y.; Lee, J.W.; Kim, S. ZEB2-Sp1 cooperation induces invasion by upregulating cadherin-11 and integrin α5 expression. Carcinogenesis 2014, 35, 302–314. [Google Scholar] [CrossRef]
- Sharma, M.; Brantley, G.J.; Vassmer, D.; Chaturvedi, G.; Baas, J.; Heuvel, G.B.V. The homeodomain protein Cux1 interacts with Grg4 to repress p27 kip1 expression during kidney development. Gene 2009, 439, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Teunissen, B.E.J.; Jansen, A.T.; van Amersfoorth, S.C.; O’Brien, T.X.; Jongsma, H.J.; Bierhuizen, M.F.A. Analysis of the rat connexin 43 proximal promoter in neonatal cardiomyocytes. Gene 2003, 322, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Thanseem, I.; Anitha, A.; Nakamura, K.; Suda, S.; Iwata, K.; Matsuzaki, H.; Ohtsubo, M.; Ueki, T.; Katayama, T.; Iwata, Y.; et al. Elevated transcription factor specificity protein 1 in autistic brains alters the expression of autism candidate genes. Biol. Psychiatry 2012, 71, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Aishworiya, R.; Valica, T.; Hagerman, R.; Restrepo, B. An Update on Psychopharmacological Treatment of Autism Spectrum Disorder. Neurotherapeutics 2022, 19, 248–262. [Google Scholar] [CrossRef] [PubMed]
- DeFilippis, M.; Wagner, K.D. Treatment of Autism Spectrum Disorder in Children and Adolescents. Psychopharmacol. Bull. 2016, 46, 18–41. [Google Scholar]
- Lopez, S.J.; Segal, D.J.; LaSalle, J.M. UBE3A: An E3 Ubiquitin Ligase With Genome-Wide Impact in Neurodevelopmental Disease. Front. Mol. Neurosci. 2019, 11, 476. [Google Scholar] [CrossRef]
- Spellman, T.; Svei, M.; Kaminsky, J.; Manzano-Nieves, G.; Liston, C. Prefrontal deep projection neurons enable cognitive flexibility via persistent feedback monitoring. Cell 2021, 184, 2750–2766.e17. [Google Scholar] [CrossRef]
- Birba, A.; García-Cordero, I.; Kozono, G.; Legaz, A.; Ibáñez, A.; Sedeño, L.; García, A.M. Losing ground: Frontostriatal atrophy disrupts language embodiment in Parkinson’s and Huntington’s disease. Neurosci. Biobehav. Rev. 2017, 80, 673–687. [Google Scholar] [CrossRef]
- Geva, S.; Schneider, L.M.; Roberts, S.; Khan, S.; Gajardo-Vidal, A.; Lorca-Puls, D.L.; Team, P.; Hope, T.M.H.; Green, D.W.; Price, C.J. Right cerebral motor areas that support accurate speech production following damage to cerebellar speech areas. Neuroimage Clin. 2021, 32, 102820. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Takahashi, K.; Liu, F.C. FOXP genes, neural development, speech and language disorders. Adv. Exp. Med. Biol. 2009, 665, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Foong, D.; Liyanage, L.; Zhou, J.; Zarrouk, A.; Ho, V.; O’Connor, M.D. Single-cell RNA sequencing predicts motility networks in purified human gastric interstitial cells of Cajal. Neurogastroenterol. Motil. 2022, 34, e14303. [Google Scholar] [CrossRef] [PubMed]
- Foong, D.; Zhou, J.; Zarrouk, A.; Ho, V.; O’Connor, M.D. Understanding the Biology of Human Interstitial Cells of Cajal in Gastrointestinal Motility. Int. J. Mol. Sci. 2020, 21, 4540. [Google Scholar] [CrossRef]
- Martire, D.; Garnier, S.; Sagnol, S.; Bourret, A.; Marchal, S.; Chauvet, N.; Guérin, A.; Forgues, D.; Berrebi, D.; Chardot, C.; et al. Phenotypic switch of smooth muscle cells in paediatric chronic intestinal pseudo-obstruction syndrome. J. Cell. Mol. Med. 2021, 25, 4028–4039. [Google Scholar] [CrossRef]
- Butler, M.G. Clinical and genetic aspects of the 15q11.2 BP1-BP2 microdeletion disorder. J. Intellect. Disabil. Res. 2017, 61, 568–579. [Google Scholar] [CrossRef]
- Sanchez-Jimeno, C.; Blanco-Kelly, F.; López-Grondona, F.; Losada-Del Pozo, R.; Moreno, B.; Rodrigo-Moreno, M.; Martinez-Cayuelas, E.; Riveiro-Alvarez, R.; Fenollar-Cortés, M.; Ayuso, C.; et al. Attention Deficit Hyperactivity and Autism Spectrum Disorders as the Core Symptoms of AUTS2 Syndrome: Description of Five New Patients and Update of the Frequency of Manifestations and Genotype-Phenotype Correlation. Genes 2021, 12, 1360. [Google Scholar] [CrossRef] [PubMed]
- Crespi, B. Genomic imprinting in the development and evolution of psychotic spectrum conditions. Biol. Rev. Camb. Philos. Soc. 2008, 83, 441–493. [Google Scholar] [CrossRef] [PubMed]
- Badcock, C.; Crespi, B. Imbalanced genomic imprinting in brain development: An evolutionary basis for the aetiology of autism. J. Evol. Biol. 2006, 19, 1007–1032. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. Increase in Developmental Disabilities among Children in the United States; Centers for Disease Control and Prevention: Atlanta, GA, USA, 2022. Available online: https://www.cdc.gov/ncbddd/developmentaldisabilities/features/increase-in-developmental-disabilities.html (accessed on 14 June 2023).
- Centers for Disease Control and Prevention. Data & Statistics on Autism Spectrum Disorder; Centers for Disease Control and Prevention: Atlanta, GA, USA, 2023. Available online: https://www.cdc.gov/ncbddd/autism/data.html (accessed on 14 June 2023).
- Ma, M.L.; Yakovenko, S.; Zhang, H.; Cheng, S.H.; Apryshko, V.; Zhavoronkov, A.; Jiang, P.; Chan, K.C.A.; Chiu, R.W.K.; Lo, Y.M.D. Fetal mitochondrial DNA in maternal plasma in surrogate pregnancies: Detection and topology. Prenat. Diagn. 2021, 41, 368–375. [Google Scholar] [CrossRef]
- Sorboni, S.G.; Moghaddam, H.S.; Jafarzadeh-Esfehani, R.; Soleimanpour, S. A Comprehensive Review on the Role of the Gut Microbiome in Human Neurological Disorders. Clin. Microbiol. Rev. 2022, 35, e0033820. [Google Scholar] [CrossRef] [PubMed]
- Okashita, N.; Tachibana, M. Transcriptional Regulation of the Y-Linked Mammalian Testis-Determining Gene SRY. Sex. Dev. 2021, 15, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Hiort, O. The differential role of androgens in early human sex development. BMC Med. 2013, 11, 152. [Google Scholar] [CrossRef] [PubMed]
- Cannarella, R.; Crafa, A.; Barbagallo, F.; Lundy, S.D.; La Vignera, S.; Condorelli, R.A.; Calogero, A.E. H19 Sperm Methylation in Male Infertility: A Systematic Review and Meta-Analysis. Int. J. Mol. Sci. 2023, 24, 7224. [Google Scholar] [CrossRef] [PubMed]
- Hvidtjørn, D.; Grove, J.; Schendel, D.; Schieve, L.A.; Sværke, C.; Ernst, E.; Thorsen, P. Risk of autism spectrum disorders in children born after assisted conception: A population-based follow-up study. J. Epidemiol. Community Health 2011, 65, 497–502. [Google Scholar] [CrossRef]
- Lehti, V.; Brown, A.S.; Gissler, M.; Rihko, M.; Suominen, A.; Sourander, A. Autism spectrum disorders in IVF children: A national case–control study in Finland. Hum. Reprod. 2013, 28, 812–818. [Google Scholar] [CrossRef]
- Farrell, A.P. How the concavity of reproduction/survival trade-offs impacts the evolution of life history strategies. J. Biol. Dyn. 2021, 15 (Suppl. 1), S134–S167. [Google Scholar] [CrossRef]
- Hawkes, K. Somatic maintenance/reproduction tradeoffs and human evolution. Behav. Brain Sci. 2022, 45, e138. [Google Scholar] [CrossRef]
- Franchini, L.F.; Pollard, K.S. Human evolution: The non-coding revolution. BMC Biol. 2017, 15, 89. [Google Scholar] [CrossRef]
- James, W.P.T.; Johnson, R.J.; Speakman, J.R.; Wallace, D.C.; Frühbeck, G.; Iversen, P.O.; Stover, P.J. Nutrition and its role in human evolution. J. Intern. Med. 2019, 285, 533–549. [Google Scholar] [CrossRef]
- Apicella, C.L.; Marlowe, F.W.; Fowler, J.H.; Christakis, N.A. Social networks and cooperation in hunter-gatherers. Nature 2012, 481, 497–501. [Google Scholar] [CrossRef] [PubMed]
- Braunschweig, D.; Van de Water, J. Maternal Autoantibodies in Autism. Arch. Neurol. 2012, 69, 693–699. [Google Scholar] [CrossRef] [PubMed]
- McLellan, J.; Kim, D.H.J.; Bruce, M.; Ramirez-Celis, A.; Van de Water, J. Maternal Immune Dysregulation and Autism–Understanding the Role of Cytokines, Chemokines and Autoantibodies. Front. Psychiatry 2022, 13, 834910. [Google Scholar] [CrossRef]
- Mazón-Cabrera, R.; Liesenborgs, J.; Brône, B.; Vandormael, P.; Somers, V. Novel maternal autoantibodies in autism spectrum disorder: Implications for screening and diagnosis. Front. Neurosci. 2023, 17, 1067833. [Google Scholar] [CrossRef]
- Dudova, I.; Horackova, K.; Hrdlicka, M.; Balastik, M. Can Maternal Autoantibodies Play an Etiological Role in ASD Development? Neuropsychiatr. Dis. Treat. 2020, 16, 1391–1398. [Google Scholar] [CrossRef]
- Maenner, M.J. Prevalence and characteristics of autism spectrum disorder among children aged 8 years—Autism and developmental disabilities monitoring network, 11 Sites, United States, 2018. MMWR Surveill. Summ. 2021, 70, 1–16. [Google Scholar] [CrossRef]
- Zeidan, J.; Fombonne, E.; Scorah, J.; Ibrahim, A.; Durkin, M.S.; Saxena, S.; Yusuf, A.; Shih, A.; Elsabbagh, M. Global prevalence of autism: A systematic review update. Autism Res. 2022, 15, 778–790. [Google Scholar] [CrossRef] [PubMed]
- Russell, G.; Stapley, S.; Newlove-Delgado, T.; Salmon, A.; White, R.; Warren, F.; Pearson, A.; Ford, T. Time trends in autism diagnosis over 20 years: A UK population-based cohort study. J. Child. Psychol. Psychiatry 2022, 63, 674–682. [Google Scholar] [CrossRef]
- Modabbernia, A.; Velthorst, E.; Reichenberg, A. Environmental risk factors for autism: An evidence-based review of systematic reviews and meta-analyses. Mol. Autism 2017, 8, 13. [Google Scholar] [CrossRef]
- Nguyen, A.; Rauch, T.A.; Pfeifer, G.P.; Hu, V.W. Global methylation profiling of lymphoblastoid cell lines reveals epigenetic contributions to autism spectrum disorders and a novel autism candidate gene, RORA, whose protein product is reduced in autistic brain. FASEB J. 2010, 24, 3036–3051. [Google Scholar] [CrossRef]
- Kumsta, R.; Hummel, E.; Chen, F.S.; Heinrichs, M. Epigenetic regulation of the oxytocin receptor gene: Implications for behavioral neuroscience. Front. Neurosci. 2013, 7, 83. [Google Scholar] [CrossRef]
- Sharma, R.; Frasch, M.G.; Zelgert, C.; Zimmermann, P.; Fabre, B.; Wilson, R.; Waldenberger, M.; MacDonald, J.W.; Bammler, T.K.; Lobmaier, S.M.; et al. Maternal-fetal stress and DNA methylation signatures in neonatal saliva: An epigenome-wide association study. Clin. Epigenetics 2022, 14, 87. [Google Scholar] [CrossRef]
- Williams, L.A.; LaSalle, J.M. Future Prospects for Epigenetics in Autism Spectrum Disorder. Mol. Diagn. Ther. 2022, 26, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, K.; Ushijima, T. Diagnostic and therapeutic applications of epigenetics. Jpn. J. Clin. Oncol. 2005, 35, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Tao, F.; Zhang, Z. Editorial: Epigenetic drugs and therapeutic resistance for epithelial malignancies. Front. Pharmacol. 2023, 14, 1208518. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Gene ID | Gene Name | GIFtS |
|---|---|---|
| ZEB1 | Zinc Finger E-Box Binding Homeobox 1 | 56 |
| PBX1 | PBX Homeobox 1 | 55 |
| NKX2-1 | NK2 Homeobox 1 | 54 |
| ZEB2 | Zinc Finger E-Box Binding Homeobox 2 | 54 |
| OTX2 | Orthodenticle Homeobox 2 | 53 |
| NKX2-5 | NK2 Homeobox 5 | 52 |
| SATB2 | SATB Homeobox 2 | 52 |
| CUX1 | Cut Like Homeobox 1 | 52 |
| EMX2 | Empty Spiracles Homeobox 2 | 51 |
| ARX | Aristaless Related Homeobox | 50 |
| SIX3 | SIX Homeobox 3 | 50 |
| ADNP | Activity-Dependent Neuroprotector Homeobox | 50 |
| DLX5 | Distal-Less Homeobox 5 | 50 |
| LMX1B | LIM Homeobox Transcription Factor 1 β | 50 |
| Drug Class | Mechanism of Action | Clinical Applications in Pediatric Disorders | Targeted Behavioral Indices | Individual Side Effects |
|---|---|---|---|---|
| Risperidone (Atypical Antipsychotic) | D2, 5HT2A Receptor Antagonist | ASD | Behavioral Irritability, Stereotyped Behaviors | ↑ Appetite, Weight Gain |
| Aripiprazole (Atypical Antipsychotic) | Partial D2 Receptor Antagonist. Partial 5HT1A Receptor Agonist | ASD | Behavioral Irritability | ↑ Appetite, Weight Gain |
| Haloperidol (Typical Antipsychotic) | D2 Receptor Antagonist | ASD | Behavioral Hyperactivity | ↑ Extrapyramidal Symptoms (Dystonia) |
| Sertraline (Antidepressant and Antianxiety) | 5HT Reuptake Inhibitor | ASD with Fragile X Syndrome | Lexical and Semantic Indices | ↑↓ Restlessness and Hyperactivity |
| Methylphenidate (Psychoactive) | NE and DA Reuptake Inhibitors | ASD with ADHD | Hyperactivity and Attention Deficits | ↓ Appetite, Abdominal Pain, Insomnia |
| Examples of Antigen Combination | % Found in Mothers of Children with ASD | % Found in Mothers of Children without ASD |
|---|---|---|
| LDH + YBX1 | 2% | 0% |
| YBX1 + CRMP2 | 5% | 0.6% |
| LDH + GDA + STIP1 | 5% | 0.6% |
| LDH + CRMP1 + STIP1 | 5% | 0% |
| STIP1 + CRMP1 + GDA | 7% | 0.6% |
| LDH + CRMP1 + STIP1 + GDA | 2% | 0% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Torres, G.; Mourad, M.; Iqbal, S.; Moses-Fynn, E.; Pandita, A.; Siddhartha, S.S.; Sood, R.A.; Srinivasan, K.; Subbaiah, R.T.; Tiwari, A.; et al. Conceptualizing Epigenetics and the Environmental Landscape of Autism Spectrum Disorders. Genes 2023, 14, 1734. https://doi.org/10.3390/genes14091734
Torres G, Mourad M, Iqbal S, Moses-Fynn E, Pandita A, Siddhartha SS, Sood RA, Srinivasan K, Subbaiah RT, Tiwari A, et al. Conceptualizing Epigenetics and the Environmental Landscape of Autism Spectrum Disorders. Genes. 2023; 14(9):1734. https://doi.org/10.3390/genes14091734
Chicago/Turabian StyleTorres, German, Mervat Mourad, Saba Iqbal, Emmanuel Moses-Fynn, Ashani Pandita, Shriya S. Siddhartha, Riya A. Sood, Kavya Srinivasan, Riya T. Subbaiah, Alisha Tiwari, and et al. 2023. "Conceptualizing Epigenetics and the Environmental Landscape of Autism Spectrum Disorders" Genes 14, no. 9: 1734. https://doi.org/10.3390/genes14091734