
Complex Autism Spectrum Disorder in a Patient with a Novel De Novo Heterozygous MYT1L Variant
 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Recruitment and Data Collection
2.2. Whole Genome Sequencing and Variant Analysis
2.3. Phenotypic Comparisons
3. Results
3.1. Clinical History and Phenotype
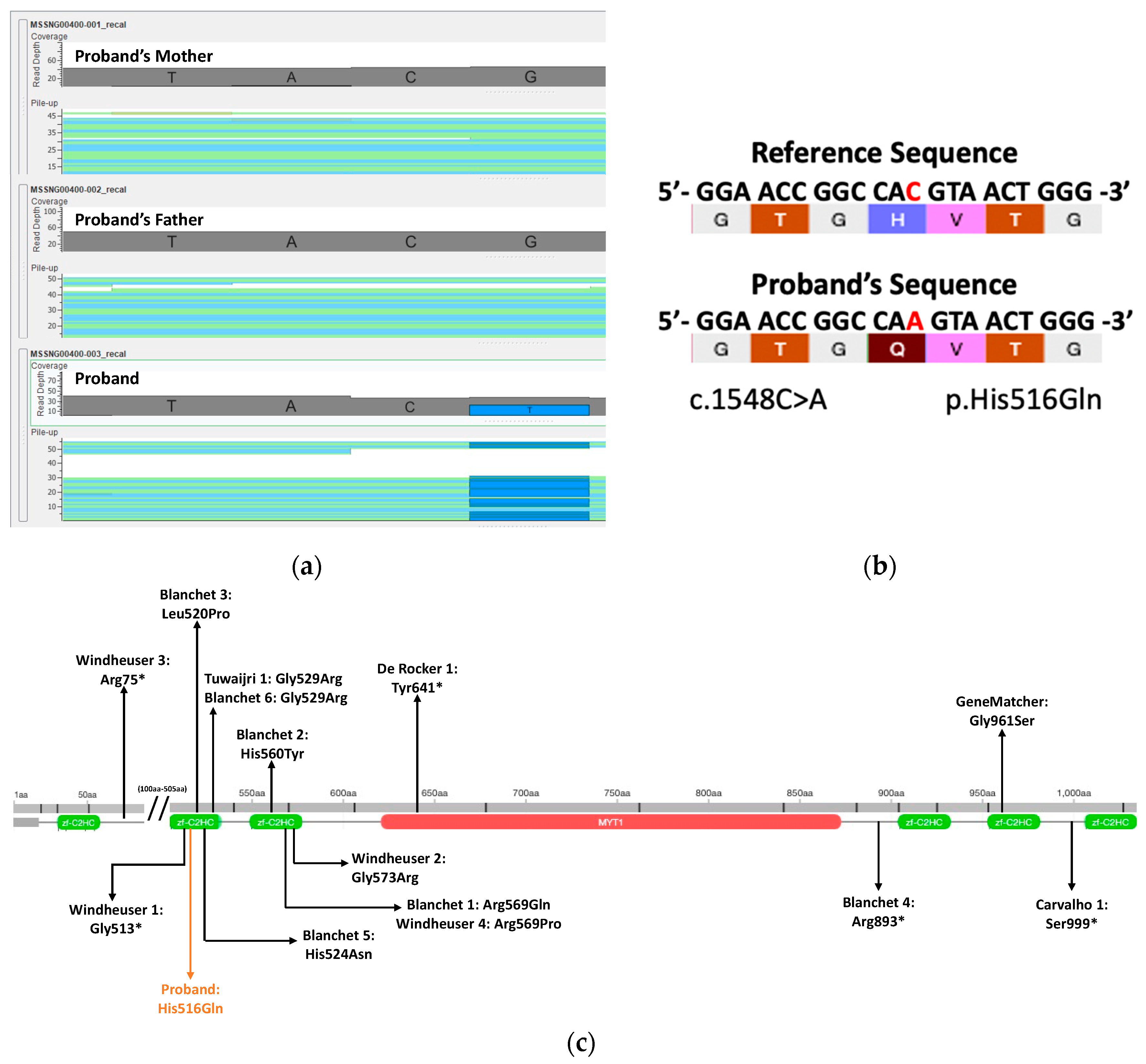
3.2. Whole Genome Sequencing and Variant Analysis Findings
3.3. Phenotypic Comparisons of Patients with De Novo SNV MYT1L Variants
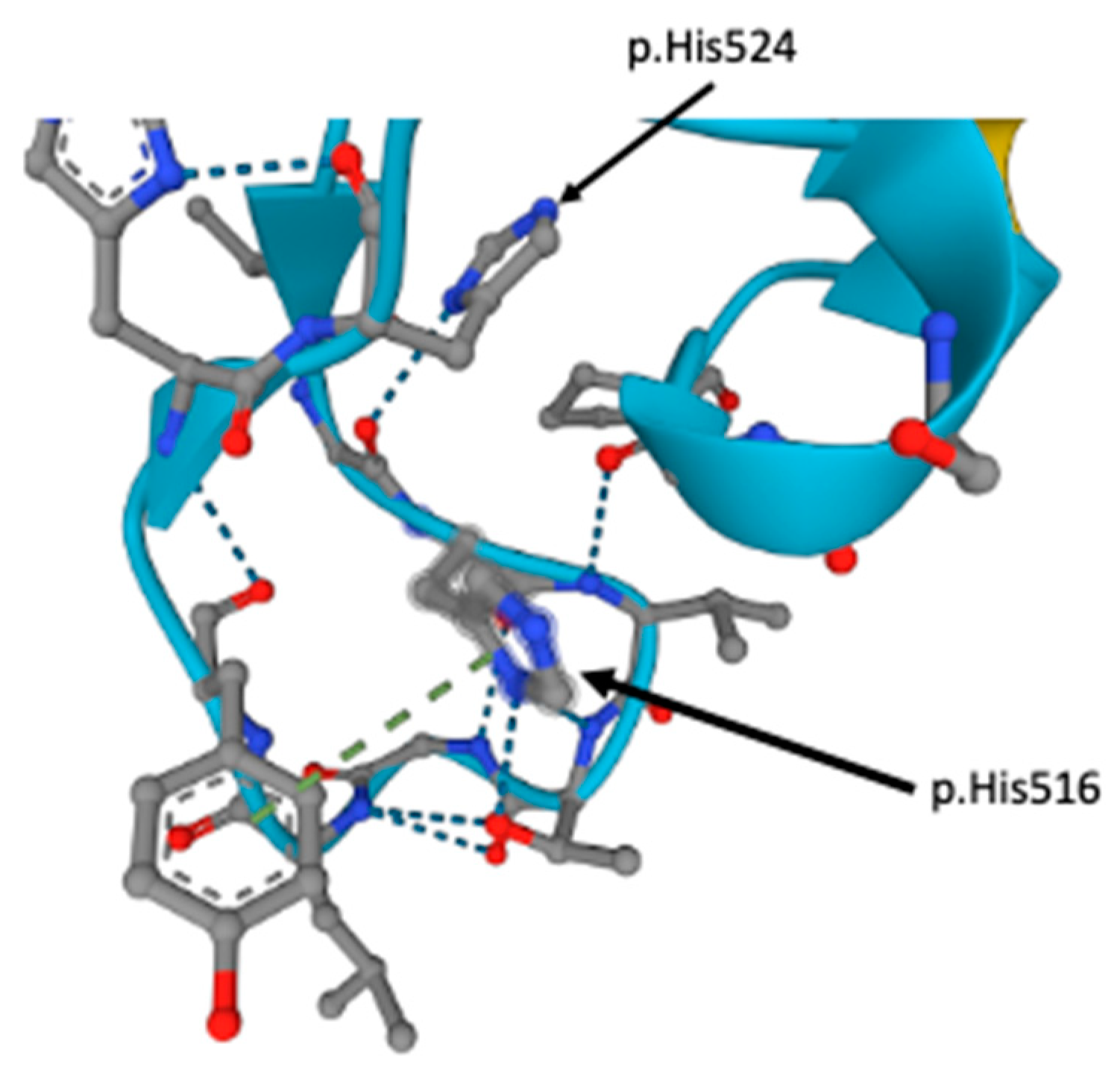
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders; DSM Library; American Psychiatric Association: Washington, DC, USA, 2013; ISBN 0-89042-555-8. [Google Scholar]
- Public Health Agency of Canada. Autism Spectrum Disorder: Highlights from the 2019 Canadian Health Survey on Children and Youth. Available online: https://www.canada.ca/en/public-health/services/publications/diseases-conditions/autism-spectrum-disorder-canadian-health-survey-children-youth-2019.html (accessed on 5 April 2023).
- Gilbert, J.; Man, H.-Y. Fundamental Elements in Autism: From Neurogenesis and Neurite Growth to Synaptic Plasticity. Front. Cell. Neurosci. 2017, 11, 359. [Google Scholar] [CrossRef] [PubMed]
- Sanders, S.J. First Glimpses of the Neurobiology of Autism Spectrum Disorder. Curr. Opin. Genet. Dev. 2015, 33, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Ayhan, F.; Konopka, G. Regulatory Genes and Pathways Disrupted in Autism Spectrum Disorders. Prog. Neuropsychopharmacol. Biol. Psychiatry 2019, 89, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Yu, V.C.; Buchholz, F.; O’Connell, S.; Rhodes, S.J.; Candeloro, C.; Xia, Y.-R.; Lusis, A.J.; Rosenfeld, M.G. A Novel Family of Cys-Cys, His-Cys Zinc Finger Transcription Factors Expressed in Developing Nervous System and Pituitary Gland (∗). J. Biol. Chem. 1996, 271, 10723–10730. [Google Scholar] [CrossRef]
- Besold, A.N.; Michel, S.L.J. Neural Zinc Finger Factor/Myelin Transcription Factor Proteins: Metal Binding, Fold, and Function. Biochemistry 2015, 54, 4443–4452. [Google Scholar] [CrossRef]
- Kim, J.G.; Armstrong, R.C.; Agoston, D.V.; Robinsky, A.; Wiese, C.; Nagle, J.; Hudson, L.D. Myelin transcription factor 1 (Myt1) of the oligodendrocyte lineage, along with a closely related CCHC zinc finger, is expressed in developing neurons in the mammalian central nervous system. J. Neurosci. Res. 1997, 50, 272–290. [Google Scholar] [CrossRef]
- Rocker, N.D.; Vergult, S.; Koolen, D.; Jacobs, E.; Hoischen, A.; Zeesman, S.; Bang, B.; Béna, F.; Bockaert, N.; Bongers, E.M.; et al. Refinement of the Critical 2p25.3 Deletion Region: The Role of MYT1L in Intellectual Disability and Obesity. Genet. Med. 2015, 17, 460–466. [Google Scholar] [CrossRef]
- Chen, J.; Yen, A.; Florian, C.P.; Dougherty, J.D. MYT1L in the Making: Emerging Insights on Functions of a Neurodevelopmental Disorder Gene. Transl. Psychiatry 2022, 12, 292. [Google Scholar] [CrossRef]
- Vierbuchen, T.; Ostermeier, A.; Pang, Z.P.; Kokubu, Y.; Südhof, T.C.; Wernig, M. Direct Conversion of Fibroblasts to Functional Neurons by Defined Factors. Nature 2010, 463, 1035–1041. [Google Scholar] [CrossRef]
- Kepa, A.; Martinez Medina, L.; Erk, S.; Srivastava, D.P.; Fernandes, A.; Toro, R.; Lévi, S.; Ruggeri, B.; Fernandes, C.; Degenhardt, F.; et al. Associations of the Intellectual Disability Gene MYT1L with Helix–Loop–Helix Gene Expression, Hippocampus Volume and Hippocampus Activation During Memory Retrieval. Neuropsychopharmacology 2017, 42, 2516–2526. [Google Scholar] [CrossRef]
- Mall, M.; Kareta, M.S.; Chanda, S.; Ahlenius, H.; Perotti, N.; Zhou, B.; Grieder, S.D.; Ge, X.; Drake, S.; Euong Ang, C.; et al. Myt1l Safeguards Neuronal Identity by Actively Repressing Many Non-Neuronal Fates. Nature 2017, 544, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Shao, Q.; Li, Z.; Gonzalez, G.A.; Lu, F.; Wang, D.; Pu, Y.; Huang, A.; Zhao, C.; He, C.; et al. Myt1L Promotes Differentiation of Oligodendrocyte Precursor Cells and Is Necessary for Remyelination After Lysolecithin-Induced Demyelination. Neurosci. Bull. 2018, 34, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Trost, B.; Thiruvahindrapuram, B.; Chan, A.J.S.; Engchuan, W.; Higginbotham, E.J.; Howe, J.L.; Loureiro, L.O.; Reuter, M.S.; Roshandel, D.; Whitney, J.; et al. Genomic Architecture of Autism from Comprehensive Whole-Genome Sequence Annotation. Cell 2022, 185, 4409–4427.e18. [Google Scholar] [CrossRef] [PubMed]
- De Rubeis, S.; He, X.; Goldberg, A.P.; Poultney, C.S.; Samocha, K.; Ercument Cicek, A.; Kou, Y.; Liu, L.; Fromer, M.; Walker, S.; et al. Synaptic, Transcriptional and Chromatin Genes Disrupted in Autism. Nature 2014, 515, 209–215. [Google Scholar] [CrossRef]
- Satterstrom, F.K.; Kosmicki, J.A.; Wang, J.; Breen, M.S.; Rubeis, S.D.; An, J.-Y.; Peng, M.; Collins, R.; Grove, J.; Klei, L.; et al. Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell 2020, 180, 568–584.e23. [Google Scholar] [CrossRef] [PubMed]
- Blanchet, P.; Bebin, M.; Bruet, S.; Cooper, G.M.; Thompson, M.L.; Duban-Bedu, B.; Gerard, B.; Piton, A.; Suckno, S.; Deshpande, C.; et al. MYT1L Mutations Cause Intellectual Disability and Variable Obesity by Dysregulating Gene Expression and Development of the Neuroendocrine Hypothalamus. PLoS Genet. 2017, 13, e1006957. [Google Scholar] [CrossRef]
- Windheuser, I.C.; Becker, J.; Cremer, K.; Hundertmark, H.; Yates, L.M.; Mangold, E.; Peters, S.; Degenhardt, F.; Ludwig, K.U.; Zink, A.M.; et al. Nine Newly Identified Individuals Refine the Phenotype Associated with MYT1L Mutations. Am. J. Med. Genet. A 2020, 182, 1021–1031. [Google Scholar] [CrossRef]
- Evans, D.R.; Qiao, Y.; Trost, B.; Calli, K.; Martell, S.; Jones, S.J.M.; Scherer, S.W.; Lewis, M.E.S. Complex Autism Spectrum Disorder with Epilepsy, Strabismus and Self-Injurious Behaviors in a Patient with a De Novo Heterozygous POLR2A Variant. Genes 2022, 13, 470. [Google Scholar] [CrossRef]
- Dhaliwal, J.; Qiao, Y.; Calli, K.; Martell, S.; Race, S.; Chijiwa, C.; Glodjo, A.; Jones, S.; Rajcan-Separovic, E.; Scherer, S.W.; et al. Contribution of Multiple Inherited Variants to Autism Spectrum Disorder (ASD) in a Family with 3 Affected Siblings. Genes 2021, 12, 1053. [Google Scholar] [CrossRef]
- Ng, P.C.; Henikoff, S. SIFT: Predicting Amino Acid Changes That Affect Protein Function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting Functional Effect of Human Missense Mutations Using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 76, 7.20.1–7.20.41. [Google Scholar] [CrossRef] [PubMed]
- Steinhaus, R.; Proft, S.; Schuelke, M.; Cooper, D.N.; Schwarz, J.M.; Seelow, D. MutationTaster2021. Nucleic Acids Res. 2021, 49, W446–W451. [Google Scholar] [CrossRef] [PubMed]
- Reva, B.; Antipin, Y.; Sander, C. Determinants of Protein Function Revealed by Combinatorial Entropy Optimization. Genome Biol. 2007, 8, R232. [Google Scholar] [CrossRef] [PubMed]
- Reva, B.; Antipin, Y.; Sander, C. Predicting the Functional Impact of Protein Mutations: Application to Cancer Genomics. Nucleic Acids Res. 2011, 39, e118. [Google Scholar] [CrossRef] [PubMed]
- Rogers, M.F.; Shihab, H.A.; Mort, M.; Cooper, D.N.; Gaunt, T.R.; Campbell, C. FATHMM-XF: Accurate Prediction of Pathogenic Point Mutations via Extended Features. Bioinformatics 2018, 34, 511–513. [Google Scholar] [CrossRef] [PubMed]
- Ioannidis, N.M.; Rothstein, J.H.; Pejaver, V.; Middha, S.; McDonnell, S.K.; Baheti, S.; Musolf, A.; Li, Q.; Holzinger, E.; Karyadi, D.; et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the Deleteriousness of Variants throughout the Human Genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef]
- Schaaf, C.P.; Betancur, C.; Yuen, R.K.C.; Parr, J.R.; Skuse, D.H.; Gallagher, L.; Bernier, R.A.; Buchanan, J.A.; Buxbaum, J.D.; Chen, C.-A.; et al. A Framework for an Evidence-Based Gene List Relevant to Autism Spectrum Disorder. Nat. Rev. Genet. 2020, 21, 367–376. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively Expanding the Structural Coverage of Protein-Sequence Space with High-Accuracy Models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Wildeman, M.; van Ophuizen, E.; den Dunnen, J.T.; Taschner, P.E.M. Improving Sequence Variant Descriptions in Mutation Databases and Literature Using the Mutalyzer Sequence Variation Nomenclature Checker. Hum. Mutat. 2008, 29, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Tuwaijri, A.A.; Alfadhel, M. MYT1L Mutation in a Patient Causes Intellectual Disability and Early Onset of Obesity: A Case Report and Review of the Literature. J. Pediatr. Endocrinol. Metab. 2019, 32, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, L.M.L.; D’Angelo, C.S.; Mustacchi, Z.; da Silva, I.T.; Krepischi, A.C.V.; Koiffmann, C.P.; Rosenberg, C. A Novel MYT1L Mutation in a Boy with Syndromic Obesity: Case Report and Literature Review. Obes. Res. Clin. Pract. 2021, 15, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Berkovits-Cymet, H.J.; Amann, B.T.; Berg, J.M. Solution Structure of a CCHHC Domain of Neural Zinc Finger Factor-1 and Its Implications for DNA Binding. Biochemistry 2004, 43, 898–903. [Google Scholar] [CrossRef]
- Besold, A.N.; Oluyadi, A.A.; Michel, S.L.J. Switching Metal Ion Coordination and DNA Recognition in a Tandem CCHHC-Type Zinc Finger Peptide. Inorg. Chem. 2013, 52, 4721–4728. [Google Scholar] [CrossRef]
- Michalek, J.L.; Besold, A.N.; Michel, S.L.J. Cysteine and Histidine Shuffling: Mixing and Matching Cysteine and Histidine Residues in Zinc Finger Proteins to Afford Different Folds and Function. Dalton Trans. 2011, 40, 12619–12632. [Google Scholar] [CrossRef]
- Lee, S.J.; Michel, S.L.J. Structural Metal Sites in Nonclassical Zinc Finger Proteins Involved in Transcriptional and Translational Regulation. Available online: https://pubs.acs.org/doi/pdf/10.1021/ar500182d (accessed on 7 April 2023).



{kind=link}
{kind=link}
| Tool | Converted Rankscore | Prediction |
|---|---|---|
| SIFT | 0.912 | Damaging |
| Polyphen2HVAR | 0.97 | Probably damaging |
| MutationTaster | 0.588 | Damaging |
| MutationAssessor | 0.906 | Predicted functional medium |
| FATHMM | 0.605 | Tolerated |
| REVEL | 0.620 | Likely causing disease |
| Phenotypic Feature | Total Cases * | Missense Cases * | LoF Cases * | Proband |
|---|---|---|---|---|
| ASD/autism | 5/14 (36%) | 3/9 (33%) | 2/5 (40%) | present |
| ID/DD | 12/13 (92%) | 8/9 (89%) | 4/4 (100%) | present |
| Speech delay | 14/14 (100%) | 9/9 (100%) | 5/5 (100%) | present |
| Motor delay | 13/13 (100%) | 9/9 (100%) | 4/4 (100%) | present |
| Obesity/overweight | 8/12 (67%) | 4/7 (57%) | 4/5 (80%) | not present |
| Hyperphagia/polyphagia | 6/8 (75%) | 4/5 (80%) | 2/3 (67%) | not present |
| Macrocephaly | 3/13 (23%) | 2/8 (25%) | 1/5 (20%) | present |
| Dysmorphic facial features | 7/11 (64%) | 3/6 (50%) | 4/5 (80%) | present |
| Behavioral abnormalities | 5/7 (71%) | 2/4 (50%) | 3/3 (100%) | present |
| Hypotonia | 7/8 (88%) | 5/6 (83%) | 2/2 (100%) | present |
| Abnormal nervous system morphology | 2/12 (17%) | 2/9 (22%) | 0/3 (0%) | present |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yip, S.; Calli, K.; Qiao, Y.; Trost, B.; Scherer, S.W.; Lewis, M.E.S. Complex Autism Spectrum Disorder in a Patient with a Novel De Novo Heterozygous MYT1L Variant. Genes 2023, 14, 2122. https://doi.org/10.3390/genes14122122
Yip S, Calli K, Qiao Y, Trost B, Scherer SW, Lewis MES. Complex Autism Spectrum Disorder in a Patient with a Novel De Novo Heterozygous MYT1L Variant. Genes. 2023; 14(12):2122. https://doi.org/10.3390/genes14122122
Chicago/Turabian StyleYip, Silas, Kristina Calli, Ying Qiao, Brett Trost, Stephen W. Scherer, and M. E. Suzanne Lewis. 2023. "Complex Autism Spectrum Disorder in a Patient with a Novel De Novo Heterozygous MYT1L Variant" Genes 14, no. 12: 2122. https://doi.org/10.3390/genes14122122