
Occurrence of L1M Elements in Chromosomal Rearrangements Associated to Chronic Myeloid Leukemia (CML): Insights from Patient-Specific Breakpoints Characterization
 , , , , , , , , ,
, , , , , , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Samples
2.2. Selection of BM CD34+/lin- Cells
2.3. CML Cell Line
2.4. Breakpoint Coordinates
3. Results
NGS and DELLY Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
Appendix A.1. Patient Material
Appendix A.2. CML Cell Line
Appendix A.3. Breakpoint Coordinates
Appendix A.4. NGS Data Analysis
Appendix A.5. Miropeats
References
- Forbes, S.A.; Beare, D.; Gunasekaran, P.; Leung, K.; Bindal, N.; Boutselakis, H.; Ding, M.; Bamford, S.; Cole, C.; Ward, S.; et al. COSMIC: Exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015, 43, D805–D811. [Google Scholar] [CrossRef] [Green Version]
- L’abbate, A.; Cunsolo, C.L.; Macrì, E.; Iuzzolino, P.; Mecucci, C.; Doglioni, C.; Coco, M.; Muscarella, L.A.; Salati, S.; Tagliafico, E.; et al. FOXP1 and TP63 involvement in the progression of myelodysplastic syndrome with 5q- and additional cytogenetic abnormalities. BMC Cancer 2014, 14, 396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Gent, D.C.; Hoeijmakers, J.H.; Kanaar, R. Chromosomal stability and the DNA double-stranded break connection. Nat. Rev. Genet. 2001, 2, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Kolomietz, E.; Meyn, M.S.; Pandita, A.; Squire, J.A. The role of Alu repeats clusters as mediators of recurrent chromosomal aberrations in tumors. Genes Chromosomes Cancer 2002, 35, 97–112. [Google Scholar] [CrossRef] [PubMed]
- Chenais, B. Transposable elements in cancer and other human diseases. Curr. Cancer Drug Targets 2015, 15, 227–242. [Google Scholar] [CrossRef]
- Price, A.L.; Eskin, E.; Pevzner, P.A. Whole-genome analysis of Alu repeat elements reveals complex evolutionary history. Genome Res. 2004, 14, 2245–2252. [Google Scholar] [CrossRef] [Green Version]
- Onno, M.; Nakamura, T.; Hillova, J.; Hill, M. Rearrangement of the human tre oncogene by homologous recombination between Alu repeats of nucleotide sequences from two different chromosomes. Oncogene 1992, 7, 2519–2523. [Google Scholar]
- Peiris, M.N.; Li, F.; Donoghue, D.J. BCR: A promiscuous fusion partner in hematopoietic disorders. Oncotarget 2019, 10, 2738–2754. [Google Scholar] [CrossRef] [Green Version]
- Mattarucchi, E.; Guerini, V.; Rambaldi, A.; Campiotti, L.; Venco, A.; Pasquali, F.; Lo Curto, F.; Porta, G. Microhomologies and Interspersed Repeat Elements at Genomic Breakpoints in Chronic Myeloid Leukemia. Cancer 2008, 301, 288–301. [Google Scholar] [CrossRef]
- Baikie, A.G.; Court-BRWM; Buckton, K.E.; Harnden, D.G.; Jakobs, P.A.; Tough, I.M. A possible specific chromosome abnormality in human chronic myeloid leukaemia. Nature 1960, 31, 1165–1166. [Google Scholar] [CrossRef]
- Carlesso, N.; Frank, D.A.; Griffin, J.D. Tyrosyl phosphorylation and DNA binding activity of signal transducers and activators of transcription (STAT) proteins in hematopoietic cell lines transformed by Bcr/Abl. J. Exp. Med. 1996, 183, 811–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melo, J.V. BCR-ABL gene variants. Baillière’s Clin. Haematol. 1997, 10, 203–222. [Google Scholar] [CrossRef]
- Pungolino, E.; D’adda, M.; De Canal, G.; Trojani, A.; Perego, A.; Elena, C.; Lunghi, F.; Turrini, M.; Borin, L.; Iurlo, A.; et al. Nilotinib-induced bone marrow CD34+/lin-Ph+ cells early clearance in newly diagnosed CP-Chronic Myeloid Leukemia: Final report of the PhilosoPhi34 study. Eur. J. Haematol. 2021, 107, 436–448. [Google Scholar] [CrossRef] [PubMed]
- Mattarucchi, E.; Spinelli, O.; Rambaldi, A.; Pasquali, F.; Curto, F.L.; Campiotti, L.; Porta, G. Molecular monitoring of residual disease in chronic myeloid leukemia by genomic DNA compared with conventional mRNA analysis. J. Mol. Diagn. 2009, 11, 482–487. [Google Scholar] [CrossRef] [Green Version]
- Pagani, I.S.; Spinelli, O.; Mattarucchi, E.; Pirrone, C.; Pigni, D.; Amelotti, E.; Lilliu, S.; Boroni, C.; Intermesoli, T.; Giussani, U.; et al. Genomic quantitative real-time PCR proves residual disease positivity in more than 30% samples with negative mRNA-based qRT-PCR in Chronic Myeloid Leukemia. Oncoscience 2014, 1, 510–521. [Google Scholar] [CrossRef] [Green Version]
- Porta, G.; Pagani, I.S.; Pirrone, C. gDNA Q-PCR for clinical monitoring of CML. Cell Cycle 2015, 14, 3659–3660. [Google Scholar] [CrossRef]
- Rainero, A.; Angaroni, F.; D’Avila, F.; Conti, A.; Pirrone, C.; Micheloni, G.; Tararà, L.; Millefanti, G.; Maserati, E.; Valli, R.; et al. GDNA qPCR is statistically more reliable than mRNA analysis in detecting leukemic cells to monitor CML. Cell Death Dis. 2018, 9, 349. [Google Scholar] [CrossRef] [Green Version]
- Trojani, A.; Pungolino, E.; Rossi, G.; D’Adda, M.; Lodola, M.; Camillo, B.D.; Perego, A.; Turrini, M.; Orlandi, E.; Borin, L.; et al. Wide-transcriptome analysis and cellularity of bone marrow CD34+/lin- cells of patients with chronic-phase chronic myeloid leukemia at diagnosis vs. 12 months of first-line nilotinib treatment. Cancer Biomark. 2017, 21, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, R.K.; Ramakrishna, W. Transposons: Unexpected players in cancer. Gene 2022, 808, 145975. [Google Scholar] [CrossRef] [PubMed]
- Jeffs, A.R.; Benjes, S.M.; Smith, T.L.; Sowerby, S.J.; Morris, C.M. The BCR gene recombines preferentially with Alu elements in complex BCR-ABL translocations of chronic myeloid leukaemia. Hum. Mol. Genet. 1998, 7, 767–776. [Google Scholar] [CrossRef] [Green Version]
- Ross, D.M.; O’Hely, M.; Bartley, P.A.; Dang, P.; Score, J.; Goyne, J.M.; Sobrinho-Simoes, M.; Cross, N.C.; Melo, J.V.; Speed, T.P.; et al. Distribution of genomic breakpoints in chronic myeloid leukemia: Analysis of 308 patients. Leukemia 2013, 27, 2105–2107. [Google Scholar] [CrossRef] [PubMed]
- Criscione, S.W.; Theodosakis, N.; Micevic, G.; Cornish, T.C.; Burns, K.H.; Neretti, N.; Rodić, N. Genome-wide characterization of human L1 antisense promoter-driven transcripts. BMC Genom. 2016, 17, 463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Martin, B.; Alvarez, E.G.; Baez-Ortega, A.; Zamora, J.; Supek, F.; Demeulemeester, J.; Santamarina, M.; Ju, Y.S.; Temes, J.; Garcia-Souto, D.; et al. Pan-cancer analysis of whole genomes identifies driver rearrangements promoted by LINE-1 retrotransposition. Nat. Genet. 2020, 52, 306–319. [Google Scholar] [CrossRef] [Green Version]
- Ehrlich, M. DNA methylation in cancer: Too much, but also too little. Oncogene 2002, 21, 5400–5413. [Google Scholar] [CrossRef] [Green Version]
- Eden, A.; Gaudet, F.; Waghmare, A.; Jaenisch, R. Chromosomal instability and tumors promoted by DNA hypomethylation. Science 2003, 300, 455. [Google Scholar] [CrossRef] [PubMed]
- Roman-Gomez, J.; Jimenez-Velasco, A.; Agirre, X.; Castillejo, J.A.; Navarro, G.; San Jose-Eneriz, E.; Garate, L.; Cordeu, L.; Cervantes, F.; Prosper, F.; et al. Repetitive DNA hypomethylation in the advanced phase of chronic myeloid leukemia. Leuk. Res. 2008, 32, 487–490. [Google Scholar] [CrossRef]
- Engreitz, J.M.; Agarwala, V.; Mirny, L.A. Three-dimensional genome architecture influences partner selection for chromosomal translocations in human disease. PLoS ONE 2012, 7, e44196. [Google Scholar] [CrossRef] [Green Version]
- Albano, F.; Anelli, L.; Zagaria, A.; Coccaro, N.; D’Addabbo, P.; Liso, V.; Rocchi, M.; Specchia, G. Genomic segmental duplications on the basis of the t(9;22) rearrangement in chronic myeloid leukemia. Oncogene 2010, 29, 2509–2516. [Google Scholar] [CrossRef] [Green Version]
- Trojani, A.; Pungolino, E.; Dal Molin, A.; Lodola, M.; Rossi, G.; D’Adda, M.; Perego, A.; Elena, C.; Turrini, M.; Borin, L.; et al. Nilotinib interferes with cell cycle, ABC transporters and JAK-STAT signaling pathway in CD34+/lin- cells of patients with chronic phase chronic myeloid leukemia after 12 months of treatment. PLoS ONE 2019, 14, e0218444. [Google Scholar] [CrossRef] [Green Version]
- Pungolino, E.; Rossi, G.; De Canal, G.; Trojani, A.; D’adda, M.; Perego, A.; Orlandi, E.M.; Lunghi, F.; Turrini, M.; Borin, L.; et al. Nilotinib induced bone marrow CD34+/lin-Ph+ cells early clearance in newly diagnosed CP-chronic myeloid leukemia. Am. J. Hematol. 2018, 93, E162–E164. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rausch, T.; Zichner, T.; Schlattl, A.; Stütz, A.M.; Benes, V.; Korbel, J.O. DELLY: Structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 2012, 28, i333–i339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
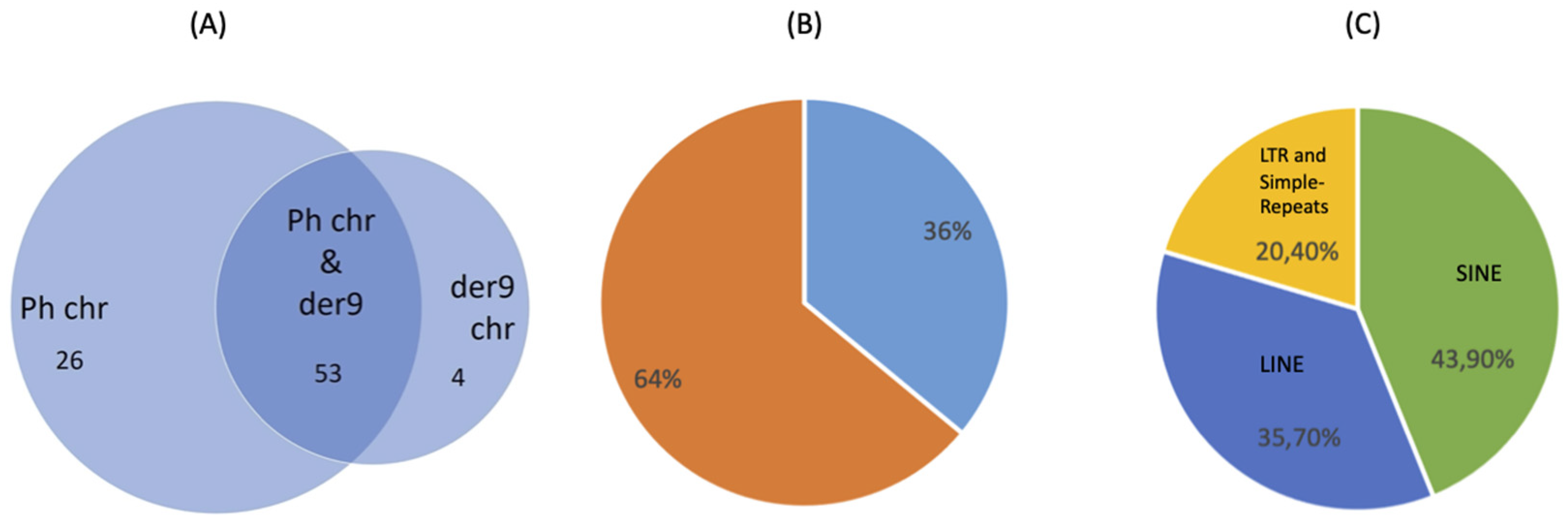
| Number of translocations | 136 | |||
| Number of BP consensus sequences | 102 | |||
| 88% micro-homologies/non-template insertion | 12% blunt | |||
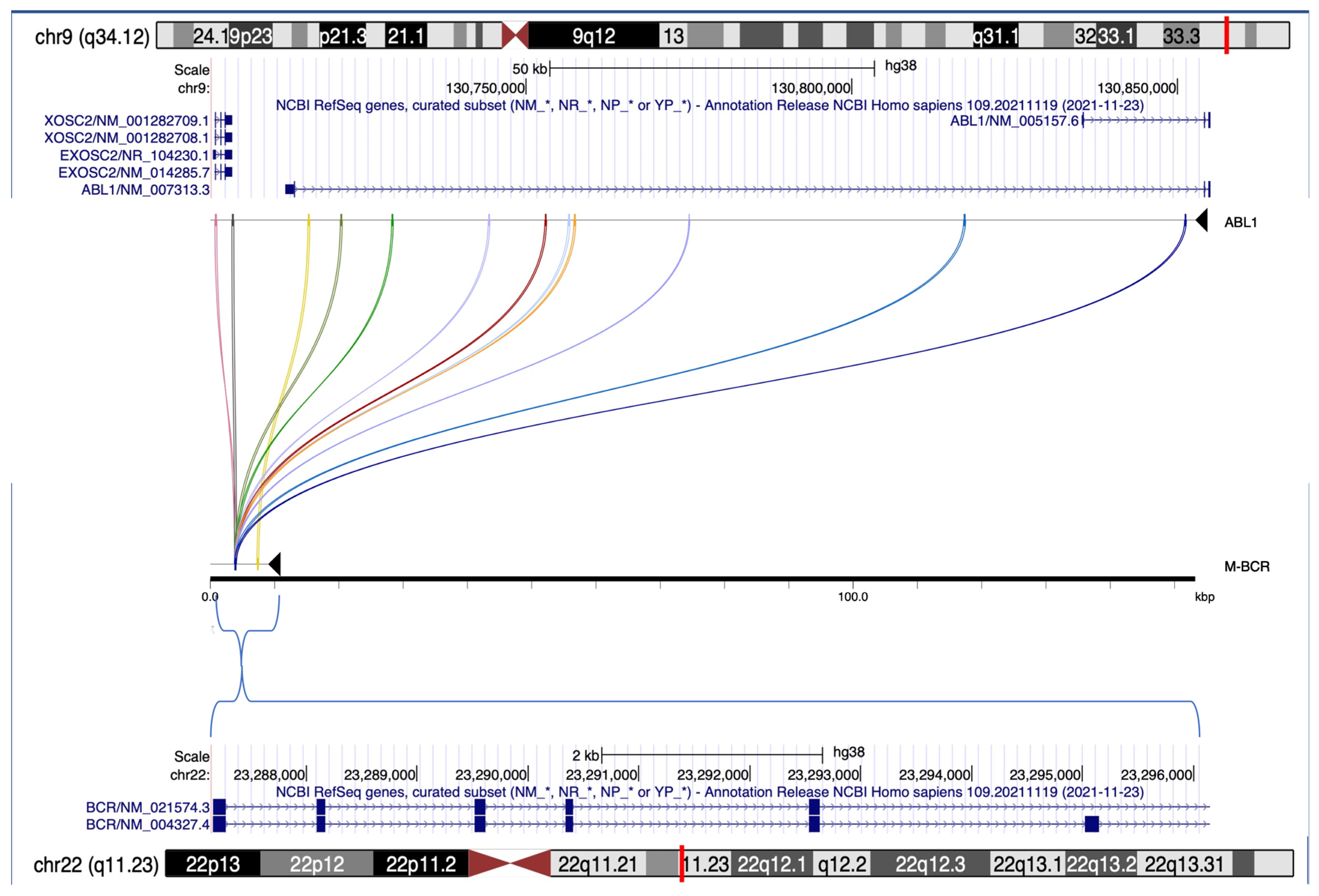
| BPs chr22 | M-bcr | m-bcr | μ-bcr | outside |
| 134 | - | - | 2 | |
| BPs chr9 | ABL1 region | outside | ||
| 128 | 8 | |||
| 36% BPs in repetitive elements | SINE | 43.9% | ||
| LINE | 35.7% | |||
| other | 20.4% | |||
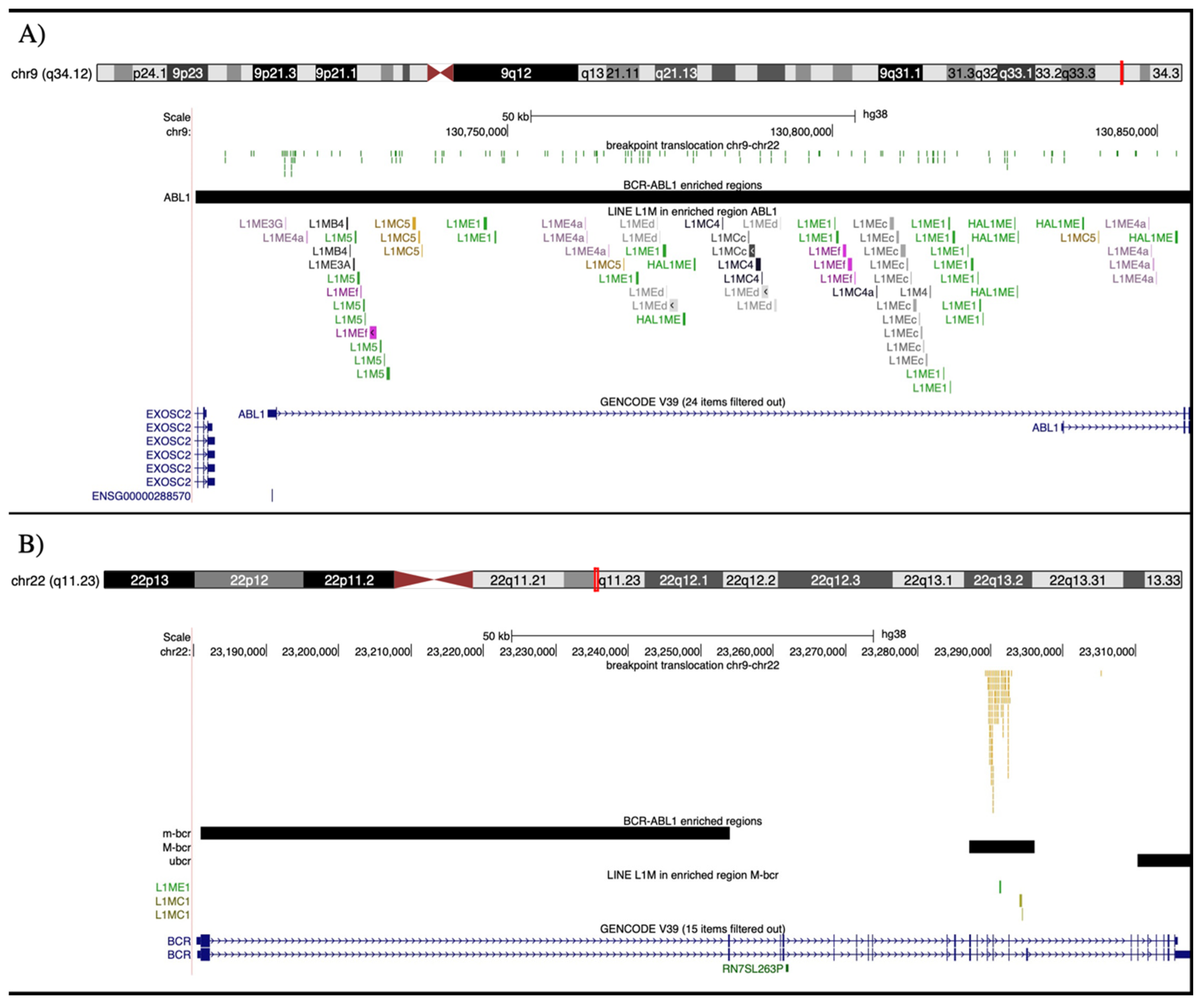
| L1M LINE enrichment (FCa > 2) | FC ABL1/RI_154 kb_mean | FC M-bcr/RI_9 kb_mean | ||
| L1ME1 | 9.5 | 10.5 | ||
| L1MCc | 12.4 | - | ||
| L1MEc | 10.6 | - | ||
| L1MEf | 8.5 | - | ||
| L1MEd | 6.6 | - | ||
| L1MC5 | 6.4 | - | ||
| L1ME4a | 4.7 | - | ||
| L1MB4 | 4.5 | - | ||
| L1M5 | 2.6 | - | ||
| L1MC4 | 2.4 | - | ||
| L1MC1 | - | 50.9 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
L’Abbate, A.; Moretti, V.; Pungolino, E.; Micheloni, G.; Valli, R.; Frattini, A.; Barcella, M.; Acquati, F.; Reinbold, R.A.; Costantino, L.; et al. Occurrence of L1M Elements in Chromosomal Rearrangements Associated to Chronic Myeloid Leukemia (CML): Insights from Patient-Specific Breakpoints Characterization. Genes 2023, 14, 1351. https://doi.org/10.3390/genes14071351
L’Abbate A, Moretti V, Pungolino E, Micheloni G, Valli R, Frattini A, Barcella M, Acquati F, Reinbold RA, Costantino L, et al. Occurrence of L1M Elements in Chromosomal Rearrangements Associated to Chronic Myeloid Leukemia (CML): Insights from Patient-Specific Breakpoints Characterization. Genes. 2023; 14(7):1351. https://doi.org/10.3390/genes14071351
Chicago/Turabian StyleL’Abbate, Alberto, Vittoria Moretti, Ester Pungolino, Giovanni Micheloni, Roberto Valli, Annalisa Frattini, Matteo Barcella, Francesco Acquati, Rolland A Reinbold, Lucy Costantino, and et al. 2023. "Occurrence of L1M Elements in Chromosomal Rearrangements Associated to Chronic Myeloid Leukemia (CML): Insights from Patient-Specific Breakpoints Characterization" Genes 14, no. 7: 1351. https://doi.org/10.3390/genes14071351