
A Carrier Female Manifesting an Unusual X-Linked Retinoschisis Phenotype Associated with the Pathogenic Variant c.266delA, p.(Tyr89LeufsTer37) in RS1, and Skewed X-Inactivation
 ,
,
Abstract
:1. Introduction
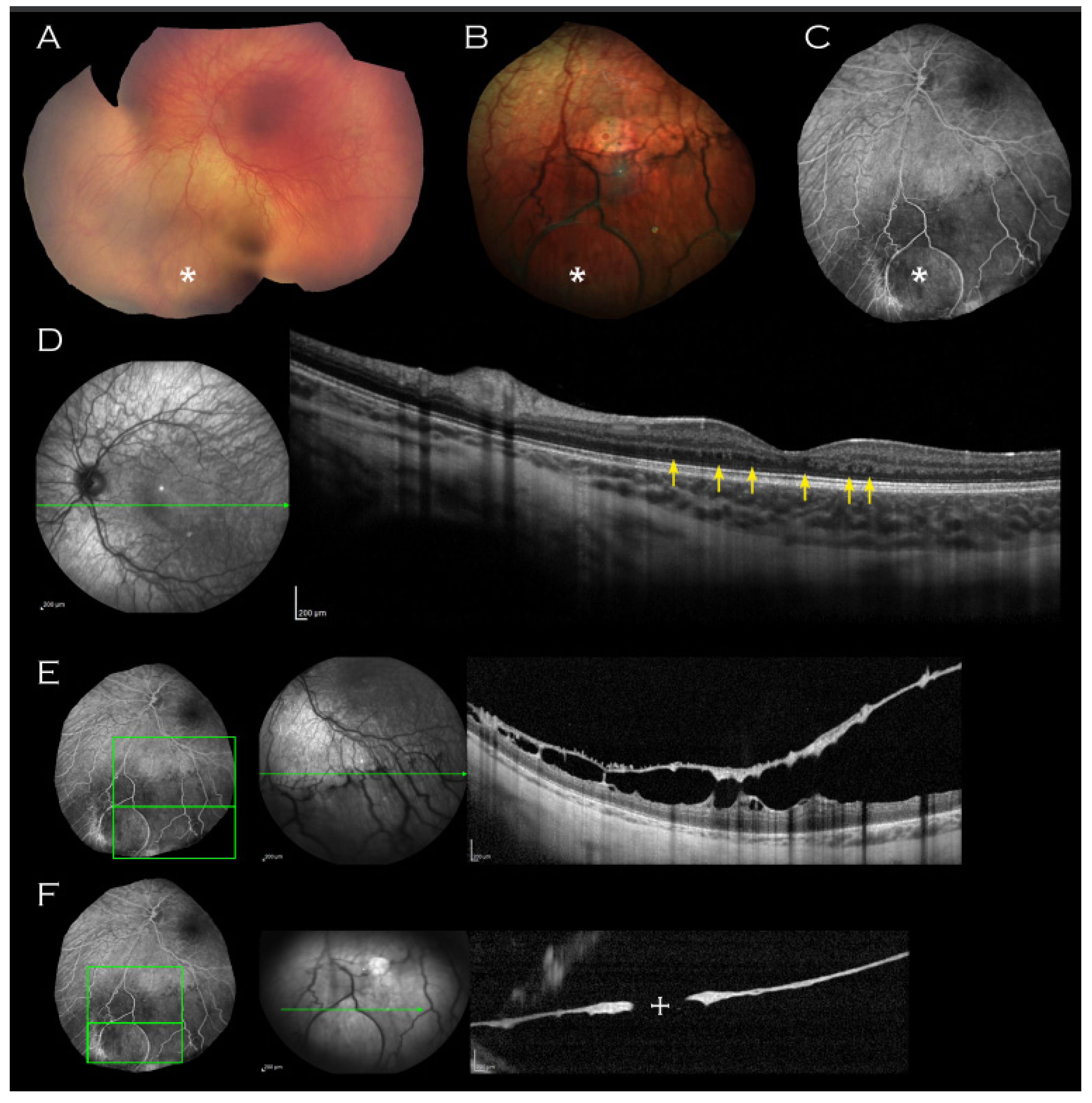
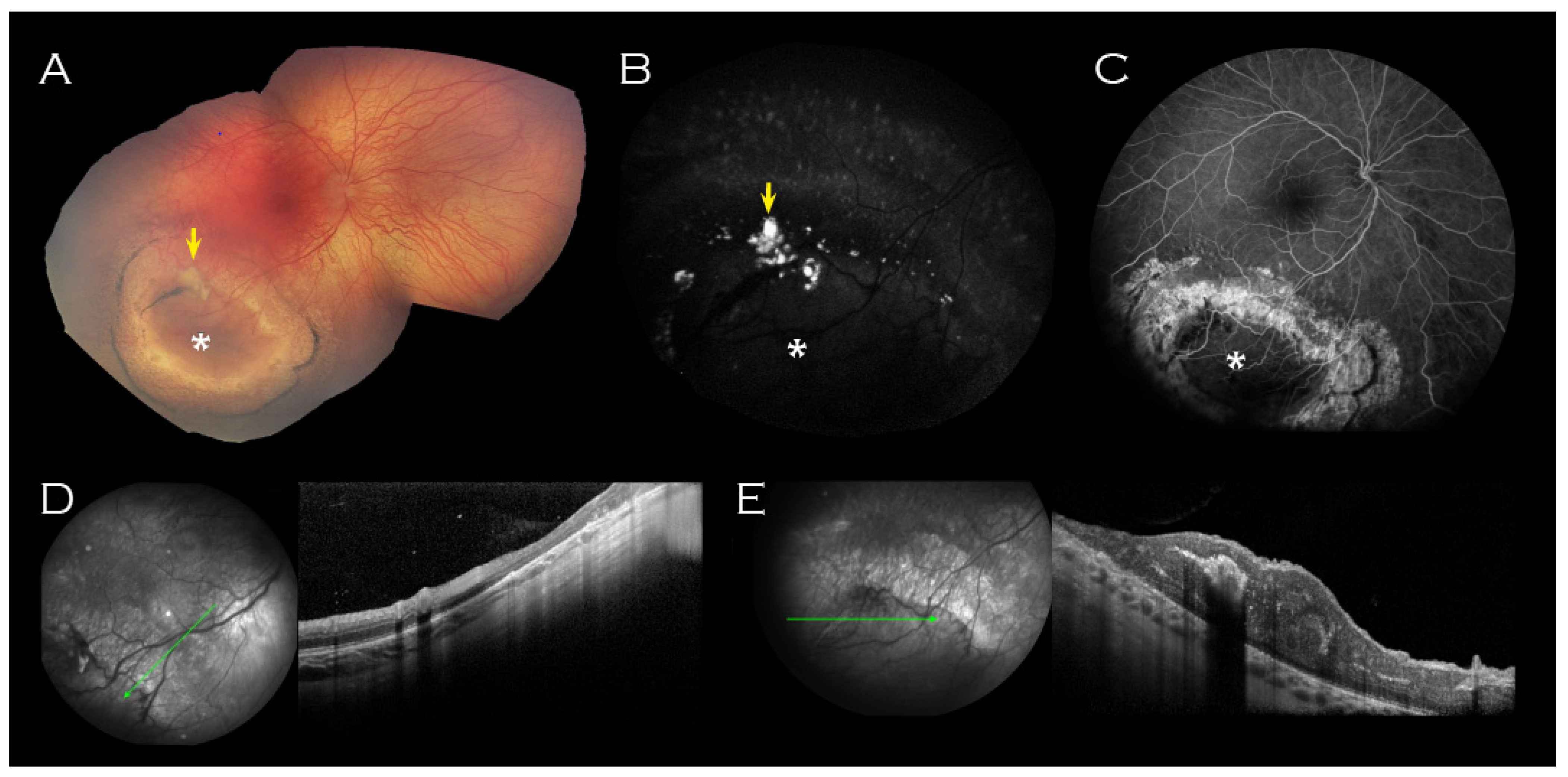
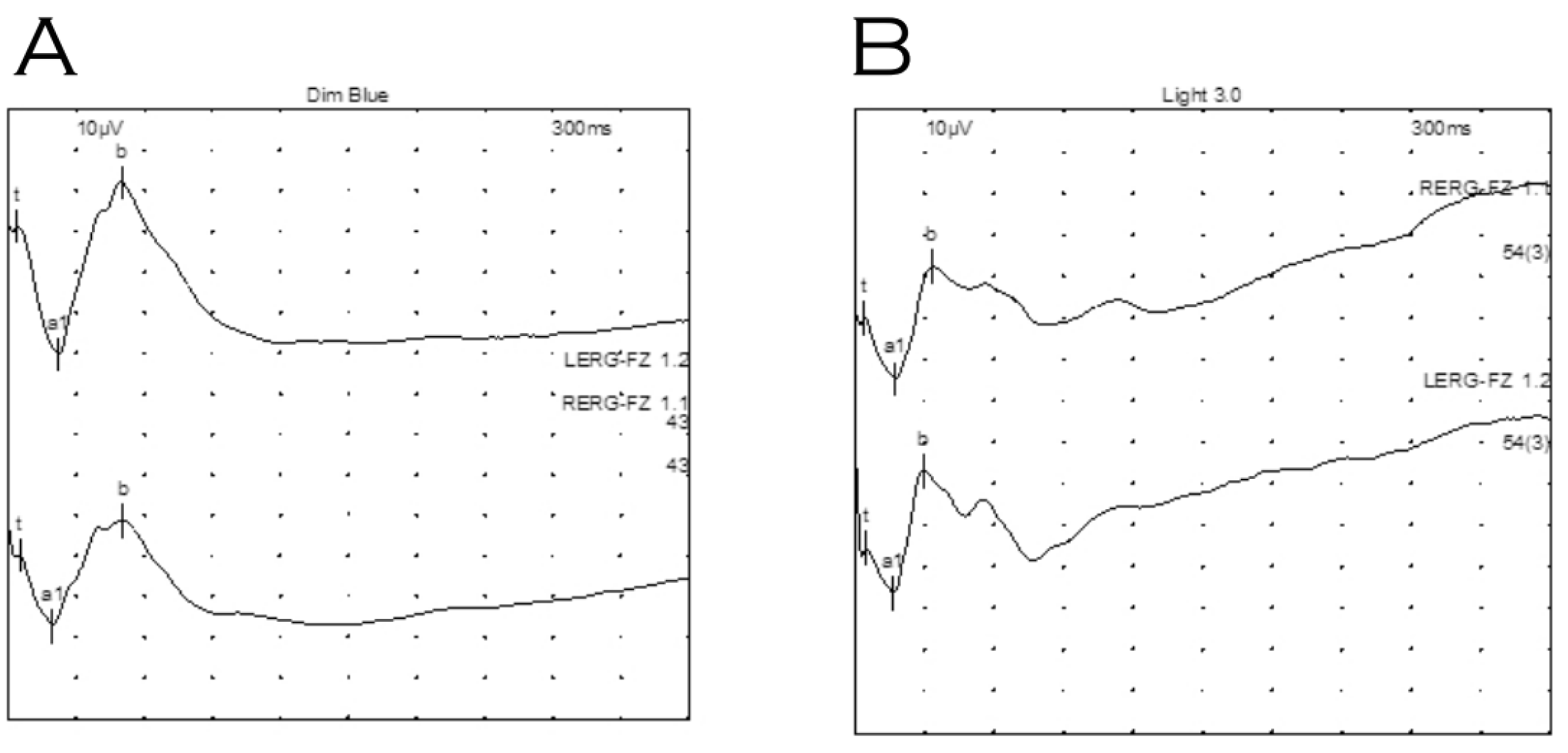
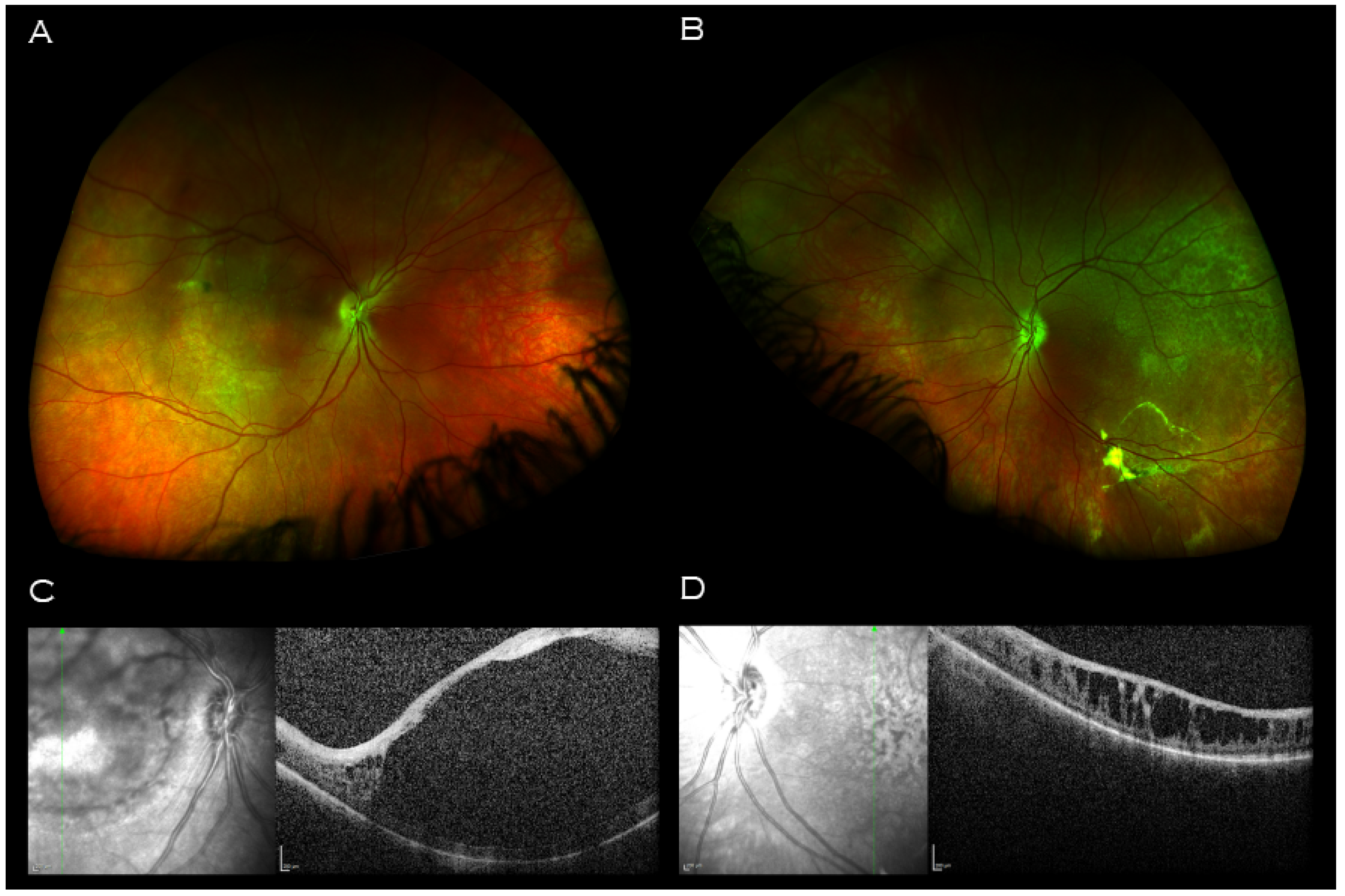
2. Case Report
3. Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sauer, C.G.; Gehrig, A.; Warneke-Wittstock, R.; Marquardt, A.; Ewing, C.C.; Gibson, A.; Lorenz, B.; Jurklies, B.; Weber, B.H. Positional cloning of the gene associated with X-linked juvenile retinoschisis. Nat. Genet. 1997, 17, 164–170. [Google Scholar] [CrossRef]
- Molday, R.S.; Kellner, U.; Weber, B.H. X-linked juvenile retinoschisis: Clinical diagnosis, genetic analysis, and molecular mechanisms. Prog. Retin. Eye Res. 2012, 31, 195–212. [Google Scholar] [CrossRef]
- Forsius, H.; Krause, U.; Helve, J.; Vuopala, V.; Mustonen, E.; Vainio-Mattila, B.; Fellman, J.; Eriksson, A.W. Visual acuity in 183 cases of X-chromosomal retinoschisis. Can. J. Ophthalmol. 1973, 8, 385–393. [Google Scholar] [PubMed]
- Sikkink, S.; Biswas, S.; Parry, N.R.A.; Stanga, P.E.; Trump, D. X-linked retinoschisis: An update. J. Med. Genet. 2007, 44, 225–232. [Google Scholar] [CrossRef] [PubMed]
- George, N.D.L.; Yates, J.R.W.; Moore, A.T. Clinical Features in Affected Males With X-Linked Retinoschisis. Arch. Ophthalmol. 1996, 114, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Alfonso-Muñoz, E.A.; Català-Mora, J.; Díaz-Cascajosa, J. X-Linked Retinoschisis without Macular Retinoschisis: A New RS1 Mutation. Ophthalmol. Retin. 2020, 4, 719. [Google Scholar] [CrossRef] [PubMed]
- Saldana, M.; Thompson, J.; Monk, E.; Trump, D.; Long, V.; Sheridan, E. X-linked retinoschisis in a female with a heterozygousRS1 missense mutation. Am. J. Med. Genet. Part A 2007, 143A, 608–609. [Google Scholar] [CrossRef] [PubMed]
- George, N.D.; Yates, J.R.; Bradshaw, K.; Moore, A.T. Infantile presentation of X linked retinoschisis. Br. J. Ophthalmol. 1995, 79, 653–657. [Google Scholar] [CrossRef] [PubMed]
- Garg, S.J.; Lee, H.C.; Grand, M.G. Bilateral Macular Detachments in X-linked Retinoschisis. Arch. Ophthalmol. 2006, 124, 1053. [Google Scholar] [CrossRef]
- Gieser, E.P.; Falls, H.F. Hereditary Retinoschisis*. Am. J. Ophthalmol. 1961, 51, 1193–1200. [Google Scholar] [CrossRef]
- Sabates, F.N. Juvenile retinoschisis. Am. J. Ophthalmol. 1966, 62, 683–688. [Google Scholar] [CrossRef]
- Wu, G.; Cotlier, E.; Brodie, S. A carrier state of X-linked juvenile retinoschisis. Ophthalmic Paediatr. Genet. 1985, 5, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, J.; Pelet, A.; Hentati, H.; Jeanpierre, M.; Briard, M.L.; Journel, H.; Munnich, A.; Dufier, J.L. Contribution to carrier detection and genetic counselling in X linked retinoschisis. J. Med. Genet. 1991, 28, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.; Seth, R. X-linked Juvenile Retinoschisis in Females and Response to Carbonic Anhydrase Inhibitors: Case Report and Review of the Literature. Semin. Ophthalmol. 2013, 28, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Feroze, A.H.; Rizvi, Z.H.; Rehman, T.-U. Consanguineous marriage resulting in homozygous occurrence of X-linked retinoschisis in girls. Am. J. Ophthalmol. 2003, 136, 767–769. [Google Scholar] [CrossRef]
- Rodríguez, F.J.; Rodríguez, A.; Mendoza-Londoño, R.; Tamayo, M.L. X-Linked Retinoschisis in Three Females from the Same Family: A Phenotype–Genotype Correlation. Retina 2005, 25, 69–74. [Google Scholar] [CrossRef]
- Saleheen, D.; Ali, A.; Khanum, S.; Ozair, M.Z.; Zaidi, M.; Sethi, M.J.; Khan, N.; Frossard, P. Molecular analysis of the XLRS1 gene in 4 females affected with X-linked juvenile retinoschisis. Can. J. Ophthalmol. 2008, 43, 596–599. [Google Scholar] [CrossRef]
- Gliem, M.; Holz, F.G.; Stohr, H.; Weber, B.H.F.; Issa, P.C. X-linked juvenile retinoschisis in a consanguineous family: Phenotypic variability and report of a homozygous female patient. Retina 2014, 34, 2472–2478. [Google Scholar] [CrossRef]
- Staffieri, S.E.; Rose, L.; Chang, A.; De Roach, J.N.; McLaren, T.L.; Mackey, D.A.; Hewitt, A.W.; Lamey, T.M. Clinical and molecular characterization of females affected by X-linked retinoschisis. Clin. Exp. Ophthalmol. 2015, 43, 643–647. [Google Scholar] [CrossRef]
- Khan, A.O.; El-Ghrably, I.A. Overlapping retinal phenotypes in a consanguineous family harboring mutations in CRB1 and RS1. Ophthalmic Genet. 2019, 40, 17–21. [Google Scholar] [CrossRef]
- Onen, M.; Zor, K.; Kucuk, E.; Iildirim, G. X-Linked Retinoschisis in Females in a Consanguineous Family: A Rare Entity. Turk. J. Ophthalmol. 2020, 50, 252–254. [Google Scholar] [CrossRef]
- Altun, A. A Female Case of X-Linked Retinoschisis with Macular Hole Bilaterally. Case Rep. Ophthalmol. Med. 2020, 2020, 8824995. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Oshika, T.; Kaji, Y.; Nose, H. Three Novel Mutations in the X-Linked Juvenile Retinoschisis (XLRS1) Gene in 6 Japanese Patients, 1 of Whom Had Turner’s Syndrome. Ophthalmic Res. 2003, 35, 295–300. [Google Scholar] [CrossRef] [PubMed]
- George, N.D.; Yates, J.R.; Moore, A.T. X linked retinoschisis. Br. J. Ophthalmol. 1995, 79, 697–702. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.; Shanks, M.; Packham, E.; Williams, J.; Haysmoore, J.; MacLaren, R.E.; Németh, A.H.; Clouston, P.; Downes, S.M. Next generation sequencing using phenotype-based panels for genetic testing in inherited retinal diseases. Ophthalmic Genet. 2020, 41, 331–337. [Google Scholar] [CrossRef]
- Al-Khuzaei, S.; Broadgate, S.; Halford, S.; Jolly, J.K.; Shanks, M.; Clouston, P.; Downes, S.M. Novel Pathogenic Sequence Variants in NR2E3 and Clinical Findings in Three Patients. Genes 2020, 11, 1288. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. ACMG Laboratory Quality Assurance Committee. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Plenge, R.M.; Hendrich, B.D.; Schwartz, C.; Arena, J.F.; Naumova, A.; Sapienza, C.; Winter, R.M.; Willard, H.F. A promoter mutation in the XIST gene in two unrelated families with skewed X-chromosome inactivation. Nat. Genet. 1997, 17, 353–356. [Google Scholar] [CrossRef]
- Fahim, A.T.; Daiger, S.P. The Role of X-Chromosome Inactivation in Retinal Development and Disease. Adv. Exp. Med. Biol. 2016, 854, 325–331. [Google Scholar]
- Carrel, L.; Willard, H.F. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature 2005, 434, 400–404. [Google Scholar] [CrossRef]
- Pegoraro, E.; Vettori, A.; Valentino, M.L.; Molon, A.; Mostacciuolo, M.L.; Howell, N.; Carelli, V. X-inactivation pattern in multiple tissues from two Leber’s hereditary optic neuropathy (LHON) patients. Am. J. Med. Genet. A 2003, 119, 37–40. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Authors | Age | Zygosity | RS1 Variant | BCVA | Macular Findings | Retinal Findings |
|---|---|---|---|---|---|---|
| Kirkby et al. 2023 | 2 years old | Heterozygous | c.266delA, p.(Tyr89LeufsTer37) | Unavailable | Thickened retinae with absence laminal structure. Mild foveal hypoplasia. Minute intraretinal hyporeflective cystic lesions; no foveal schisis. | Inferior peripheral retinoschisis (left). Inferotemporal RPE atrophy (right, with associated concentric rings of pigment enclosing a schitic-appearing area and yellow material). |
| Saldana et al. [7] | 5 years old | Heterozygous | c.305G>A; p.(Arg102Gln) | 20/30 and 20/40 | RPE changes; no foveal schisis. | Bilateral inferotemporal peripheral retinoschisis. |
| Gieser et al. 1961 [10] | 13 years old | Obligate carrier | Unavailable | 20/25 (Central scotomas) | Foveal schisis with “pigment mottling and few verrucae in sharply punched-out macular area.” | |
| Sabates 1966 [11] | 41 years old | Obligate carrier | Unavailable | Unavailable | Temporal (right) and inferotemporal (left) retinoschisis. Two areas midperipheral “pigment clumping.” | |
| Wu et al. 1985 [12] | 56 years old | Obligate carrier | Unavailable | Unavailable | Radial wrinkling around fovea, internal limiting membrane; no foveal schisis | |
| Kaplan et at 1991 [13] | Unavailable | 5 x Obligate carriers | Unavailable | Unavailable | None had macular findings | Peripheral lesions (“mild greyish-white spots or dendrite-like areas,” “aberrant zones of underdeveloped capillaries,” inferotemporal retinoschisis). Two had previous childhood cryocoagulation. |
| Ali, S et al. 2013 [14] | 54 years old | Obligate carrier | Unavailable | Unavailable | Inferior retinoschisis, outer retinal hole with lattice degeneration in one eye. | |
| Ali, A et al. 2003 [15] | Proband (unavailable) | Presumed homozygous or compound heterozygous | Unavailable | LP (Bilateral nystagmus) | Bilateral total retinal detachment. | |
| 10 years old | Presumed homozygous or compound heterozygous | Unavailable | 6/60 and 3/60 | Foveal schisis. | Bilateral longstanding inferior retinoschisis extending to inferior arcades. | |
| 5 years old | Presumed homozygous or compound heterozygous | Unavailable | 6/36 and 6/60 | Foveal schisis with pigmentary changes. | Bilateral inferior scars and retinoschisis with pigmentary changes. | |
| 1 year old | Presumed homozygous or compound heterozygous | Unavailable | Unavailable | “Macular involvement” of retinoschisis. | Bilateral retinal detachment, and retinoschisis. Secondary changes in areas of detachment, markedly atrophic retinae. | |
| Rodriguez, F.J. et al., 2005 [16] | 10 years old | Homozygous | c.639delG | 20/20 and 20/30 | Foveal schisis. | Mid-retinal cyst inferotemporal quadrant. Pale optic discs. |
| 37 years old | Homozygous | c.639delG | 20/100 and 20/400 | “Modified” foveal schisis | Previous cryotherapy inferotemporally. Pale optic discs. | |
| 37 years old | Homozygous | c.639delG | 20/60 and 20/400 | “Modified” foveal schisis | Peripheral retinoschisis. Pale optic discs. | |
| Saleheen, D. et al. 2008 [17] | 1 year old | Homozygous | c.579dupC | Unavailable | “Macular involvement” of retinoschisis. | Bilateral retinal detachment. |
| 3 years old | Homozygous | c.579dupC | LP (Bilateral nystagmus) | Bilateral complete retinal detachment. | ||
| 10 years old | Homozygous | c.579dupC | 6/60 and 3/60 | Foveal schisis radiating to inferior arcades. | ||
| 5 years old | Homozygous | c.579dupC | 6/36 and 6/60 | Foveal schisis. | Bilateral inferior scars with pigmentary changes accompanying peripheral retinoschisis. | |
| Gliem, M. et al. 2014 [18] | 59 years old | Homozygous | c.293C>A, p.(Ala98Glu) | Unavailable | Atrophic macula. | Hyperpigmentary changes and chorioretinal scarring towards periphery. Prior cryotherapy. |
| Staffieri, S.E. et al., 2015 [19] | 9 years old | Homozygous | c.304C>T (rs61752067) | Sensory nystagmus | ||
| 11 years old | Homozygous | c.304C>T (rs61752067) | Sensory nystagmus | Foveal schisis. | ||
| Khan, A.O. et al., 2019 [20] | Unavailable | Homozygous | c.304C>T p.(Arg102Trp) | “Poor since childhood” | Foveal schisis | Peripheral retinoschisis. |
| Onen, M. et al. 2020 [21] | 4 years old | Presumed homozygous or compound heterozygous | Not specified | 20/50 | Foveal schisis. | |
| 15 years old | Presumed homozygous or compound heterozygous | Not specified | 20/40 | Foveal schisis. | ||
| 17 years old | Presumed homozygous or compound heterozygous | Not specified | 20/100 | Atrophic macula with RPE changes. | ||
| Altun, A. et al. 2020 [22] | 18 years old | Presumed homozygous or compound heterozygous | Not specified | CF | Bilateral macula holes. | |
| Sato, M. et al. 2003 [23] | 29 years old | Turners syndrome | c.(522+1G>A) | 60/200 | Foveal schisis (diagnosed at 10 years old). |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kirkby, J.; Halford, S.; Shanks, M.; Moore, A.; Gait, A.; Jenkins, L.; Clouston, P.; Patel, C.K.; Downes, S.M. A Carrier Female Manifesting an Unusual X-Linked Retinoschisis Phenotype Associated with the Pathogenic Variant c.266delA, p.(Tyr89LeufsTer37) in RS1, and Skewed X-Inactivation. Genes 2023, 14, 1193. https://doi.org/10.3390/genes14061193
Kirkby J, Halford S, Shanks M, Moore A, Gait A, Jenkins L, Clouston P, Patel CK, Downes SM. A Carrier Female Manifesting an Unusual X-Linked Retinoschisis Phenotype Associated with the Pathogenic Variant c.266delA, p.(Tyr89LeufsTer37) in RS1, and Skewed X-Inactivation. Genes. 2023; 14(6):1193. https://doi.org/10.3390/genes14061193
Chicago/Turabian StyleKirkby, Jennifer, Stephanie Halford, Morag Shanks, Anthony Moore, Anthony Gait, Lucy Jenkins, Penny Clouston, Chetan K. Patel, and Susan M. Downes. 2023. "A Carrier Female Manifesting an Unusual X-Linked Retinoschisis Phenotype Associated with the Pathogenic Variant c.266delA, p.(Tyr89LeufsTer37) in RS1, and Skewed X-Inactivation" Genes 14, no. 6: 1193. https://doi.org/10.3390/genes14061193