


Genome-Wide Integrative Transcriptional Profiling Identifies Age-Associated Signatures in Dogs


Abstract
:1. Introduction
2. Materials and Methods
2.1. RNA-Sequencing Data and Analysis of Differentially Expressed Genes
2.2. Age-Dependent DNA Methylation
2.3. Gene Set Enrichment Analysis
2.4. Figure Visualization
3. Results and Discussion
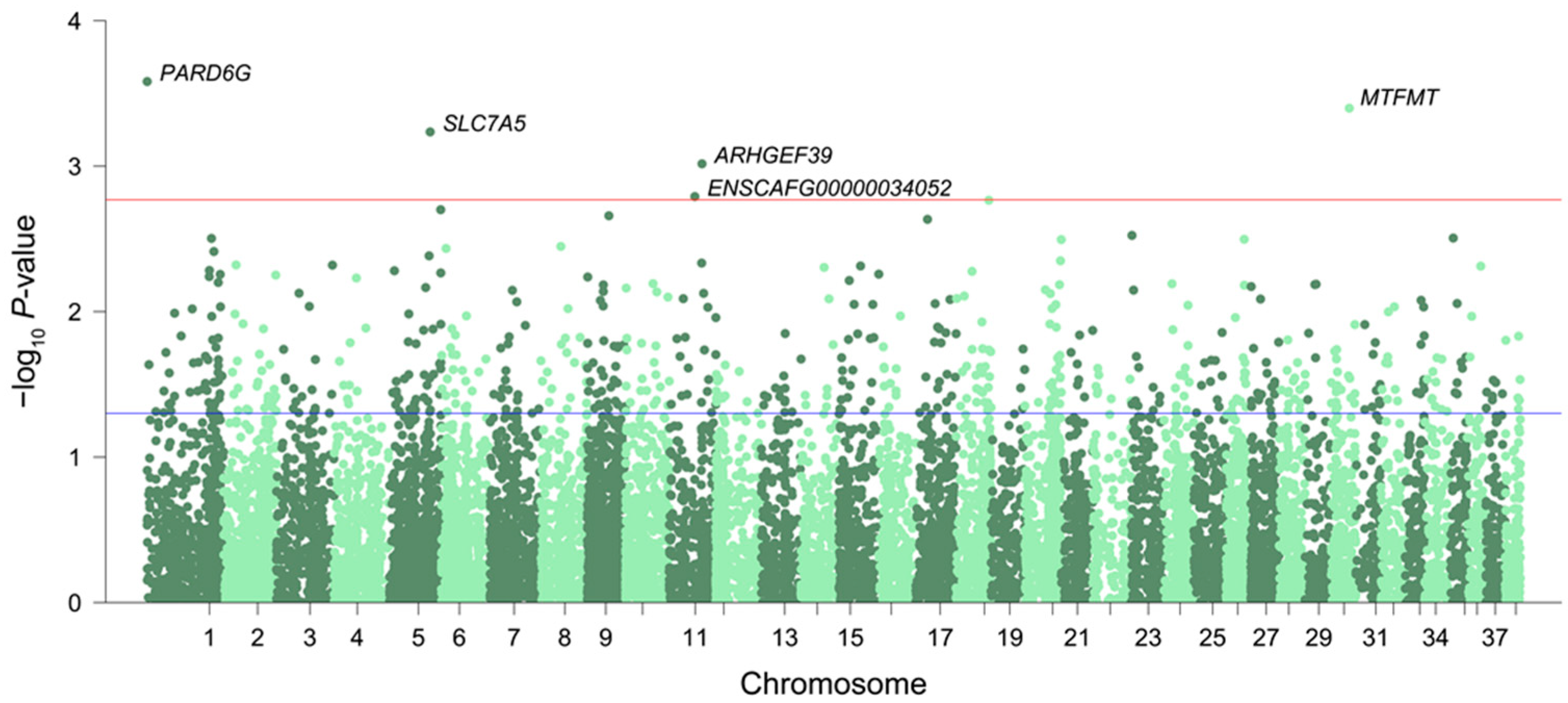
3.1. Differentially Expressed Genes and Enriched Pathways Associated with Aging
3.2. Conserved Signatures across Mammals
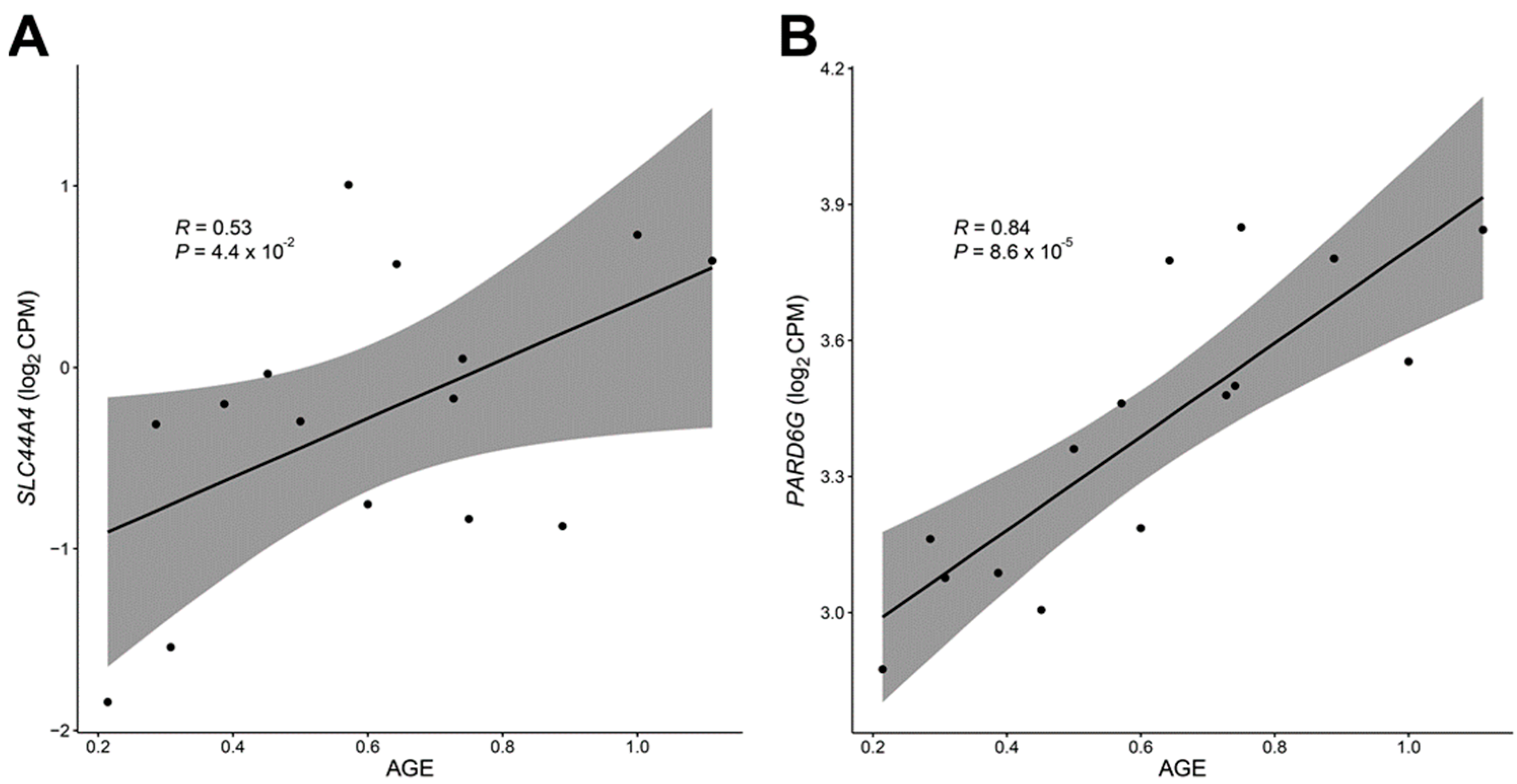
3.3. Differentially Expressed Genes with Age-Related DNA Methylation Patterns
3.4. Limitations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wayne, R.K.; Ostrander, E.A. Lessons learned from the dog genome. Trends Genet. 2007, 23, 557–567. [Google Scholar] [CrossRef] [PubMed]
- Neilson, J.C.; Hart, B.L.; Cliff, K.D.; Ruehl, W.W. Prevalence of behavioral changes associated with age-related cognitive impairment in dogs. J. Am. Vet. Med. Assoc. 2001, 218, 1787–1791. [Google Scholar] [CrossRef] [PubMed]
- Hirai, T.; Kojima, S.; Shimada, A.; Umemura, T.; Sakai, M.; Itakurat, C. Age-related changes in the olfactory system of dogs. Neuropathol. Appl. Neurobiol. 1996, 22, 531–539. [Google Scholar] [CrossRef]
- Sándor, S.; Kubinyi, E. Genetic pathways of aging and their relevance in the dog as a natural model of human aging. Front. Genet. 2019, 10, 948. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, K.M.; Greer, K.A. Why is the dog an ideal model for aging research? Exp. Gerontol. 2015, 71, 14–20. [Google Scholar] [CrossRef]
- Olson, P.; Bonnett, B.; Hedhammar, Å.; Egenvall, A. Mortality in over 350,000 Insured Swedish Dogs from 1995–2000: II. Breed-Specific Age and Survival Patterns and Relative Risk for Causes of Death. Acta Vet. Scand. 2005, 46, 121–136. [Google Scholar]
- Abadie, J.; Hédan, B.; Cadieu, E.; De Brito, C.; Devauchelle, P.; Bourgain, C.; Parker, H.G.; Vaysse, A.; Margaritte-Jeannin, P.; Galibert, F. Epidemiology, pathology, and genetics of histiocytic sarcoma in the Bernese mountain dog breed. J. Hered. 2009, 100, S19–S27. [Google Scholar] [CrossRef]
- Schaible, R.H. Genetic predisposition to purine uroliths in Dalmatian dogs. Vet. Clin. N. Am. Small Anim. Pract. 1986, 16, 127–131. [Google Scholar] [CrossRef]
- Bannasch, D.; Ling, G.; Bea, J.; Famula, T. Inheritance of urinary calculi in the Dalmatian. J. Vet. Intern. Med. 2004, 18, 483–487. [Google Scholar] [CrossRef]
- Bannasch, D.; Safra, N.; Young, A.; Karmi, N.; Schaible, R.; Ling, G. Mutations in the SLC2A9 gene cause hyperuricosuria and hyperuricemia in the dog. PLoS Genet. 2008, 4, e1000246. [Google Scholar] [CrossRef]
- Kraus, C.; Pavard, S.; Promislow, D.E. The size–life span trade-off decomposed: Why large dogs die young. Am. Nat. 2013, 181, 492–505. [Google Scholar] [CrossRef] [PubMed]
- Ruple, A.; MacLean, E.; Snyder-Mackler, N.; Creevy, K.E.; Promislow, D. Dog models of aging. Annu. Rev. Anim. Biosci. 2022, 10, 419–439. [Google Scholar] [CrossRef] [PubMed]
- Labbadia, J.; Morimoto, R.I. Repression of the heat shock response is a programmed event at the onset of reproduction. Mol. Cell 2015, 59, 639–650. [Google Scholar] [CrossRef]
- Solis, G.M.; Kardakaris, R.; Valentine, E.R.; Bar-Peled, L.; Chen, A.L.; Blewett, M.M.; McCormick, M.A.; Williamson, J.R.; Kennedy, B.; Cravatt, B.F. Translation attenuation by minocycline enhances longevity and proteostasis in old post-stress-responsive organisms. Elife 2018, 7, e40314. [Google Scholar] [CrossRef]
- Harman, D. Aging: Overview. Ann. N. Y. Acad. Sci. 2001, 928, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Pagiatakis, C.; Musolino, E.; Gornati, R.; Bernardini, G.; Papait, R. Epigenetics of aging and disease: A brief overview. Aging Clin. Exp. Res. 2021, 33, 737–745. [Google Scholar] [CrossRef]
- Jeong, H.; Wu, X.; Smith, B.; Yi, S.V. Genomic landscape of methylation islands in hymenopteran insects. Genome Biol. Evol. 2018, 10, 2766–2776. [Google Scholar] [CrossRef]
- Brunet, A.; Berger, S.L. Epigenetics of aging and aging-related disease. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2014, 69, S17–S20. [Google Scholar] [CrossRef]
- Pal, S.; Tyler, J.K. Epigenetics and aging. Sci. Adv. 2016, 2, e1600584. [Google Scholar] [CrossRef]
- De Magalhães, J.P.; Curado, J.; Church, G.M. Meta-analysis of age-related gene expression profiles identifies common signatures of aging. Bioinformatics 2009, 25, 875–881. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Berdyshev, G.; Korotaev, G.; Boiarskikh, G.; Vaniushin, B. Nucleotide composition of DNA and RNA from somatic tissues of humpback and its changes during spawning. Biokhimiia 1967, 32, 988–993. [Google Scholar] [PubMed]
- Romanov, G.A.; Vanyushin, B.F. Methylation of reiterated sequences in mammalian DNAs Effects of the tissue type, age, malignancy and hormonal induction. Biochim. Biophys. Acta (BBA) Nucleic Acids Protein Synth. 1981, 653, 204–218. [Google Scholar] [CrossRef]
- Wilson, V.L.; Jones, P.A. DNA methylation decreases in aging but not in immortal cells. Science 1983, 220, 1055–1057. [Google Scholar] [CrossRef] [PubMed]
- Wilson, V.L.; Smith, R.; Ma, S.; Cutler, R. Genomic 5-methyldeoxycytidine decreases with age. J. Biol. Chem. 1987, 262, 9948–9951. [Google Scholar] [CrossRef] [PubMed]
- Fairweather, D.S.; Fox, M.; Margison, G.P. The in vitro lifespan of MRC-5 cells is shortened by 5-azacytidine-induced demethylation. Exp. Cell Res. 1987, 168, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; Raj, K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat. Rev. Genet. 2018, 19, 371–384. [Google Scholar] [CrossRef]
- Hahn, O.; Grönke, S.; Stubbs, T.M.; Ficz, G.; Hendrich, O.; Krueger, F.; Andrews, S.; Zhang, Q.; Wakelam, M.J.; Beyer, A. Dietary restriction protects from age-associated DNA methylation and induces epigenetic reprogramming of lipid metabolism. Genome Biol. 2017, 18, 56. [Google Scholar] [CrossRef]
- Thompson, M.J.; Horvath, S.; Pellegrini, M. An epigenetic aging clock for dogs and wolves. Aging 2017, 9, 1055–1068. [Google Scholar] [CrossRef]
- Wang, T.; Ma, J.; Hogan, A.N.; Fong, S.; Licon, K.; Tsui, B.; Kreisberg, J.F.; Adams, P.D.; Carvunis, A.-R.; Bannasch, D.L. Quantitative translation of dog-to-human aging by conserved remodeling of the DNA methylome. Cell Syst. 2020, 11, 176–185.e6. [Google Scholar] [CrossRef]
- Piétu, G.; Mariage-Samson, R.; Fayein, N.-A.; Matingou, C.; Eveno, E.; Houlgatte, R.; Decraene, C.; Vandenbrouck, Y.; Tahi, F.; Devignes, M.-D. The Genexpress IMAGE knowledge base of the human brain transcriptome: A prototype integrated resource for functional and computational genomics. Genome Res. 1999, 9, 195–209. [Google Scholar] [CrossRef]
- Lowe, R.; Shirley, N.; Bleackley, M.; Dolan, S.; Shafee, T. Transcriptomics technologies. PLoS Comput. Biol. 2017, 13, e1005457. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Ham, S.; Lee, S.-J.V. Advances in transcriptome analysis of human brain aging. Exp. Mol. Med. 2020, 52, 1787–1797. [Google Scholar] [CrossRef]
- Peters, M.J.; Joehanes, R.; Pilling, L.C.; Schurmann, C.; Conneely, K.N.; Powell, J.; Reinmaa, E.; Sutphin, G.L.; Zhernakova, A.; Schramm, K. The transcriptional landscape of age in human peripheral blood. Nat. Commun. 2015, 6, 8570. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Campisi, J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2014, 69, S4–S9. [Google Scholar] [CrossRef] [PubMed]
- Cannon, L.; Zambon, A.C.; Cammarato, A.; Zhang, Z.; Vogler, G.; Munoz, M.; Taylor, E.; Cartry, J.; Bernstein, S.I.; Melov, S. Expression patterns of cardiac aging in Drosophila. Aging Cell 2017, 16, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Shavlakadze, T.; Morris, M.; Fang, J.; Wang, S.X.; Zhu, J.; Zhou, W.; Herman, W.T.; Mondragon-Gonzalez, R.; Roma, G.; Glass, D.J. Age-related gene expression signature in rats demonstrate early, late, and linear transcriptional changes from multiple tissues. Cell Rep. 2019, 28, 3263–3273.e3. [Google Scholar] [CrossRef]
- Perez-Gomez, A.; Buxbaum, J.N.; Petrascheck, M. The aging transcriptome: Read between the lines. Curr. Opin. Neurobiol. 2020, 63, 170–175. [Google Scholar] [CrossRef]
- Rangaraju, S.; Levey, D.; Nho, K.; Jain, N.; Andrews, K.; Le-Niculescu, H.; Salomon, D.; Saykin, A.; Petrascheck, M.; Niculescu, A. Mood, stress and longevity: Convergence on ANK3. Mol. Psychiatry 2016, 21, 1037–1049. [Google Scholar] [CrossRef]
- Han, Y.; Eipel, M.; Franzen, J.; Sakk, V.; Dethmers-Ausema, B.; Yndriago, L.; Izeta, A.; de Haan, G.; Geiger, H.; Wagner, W. Epigenetic age-predictor for mice based on three CpG sites. Elife 2018, 7, e37462. [Google Scholar] [CrossRef] [PubMed]
- Fahy, G.M.; Brooke, R.T.; Watson, J.P.; Good, Z.; Vasanawala, S.S.; Maecker, H.; Leipold, M.D.; Lin, D.T.; Kobor, M.S.; Horvath, S. Reversal of epigenetic aging and immunosenescent trends in humans. Aging Cell 2019, 18, e13028. [Google Scholar] [CrossRef] [PubMed]
- Wiesman, A.I.; Rezich, M.T.; O’Neill, J.; Morsey, B.; Wang, T.; Ideker, T.; Swindells, S.; Fox, H.S.; Wilson, T.W. Epigenetic markers of aging predict the neural oscillations serving selective attention. Cereb. Cortex 2020, 30, 1234–1243. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, D.L.; Barrett, T.; Benson, D.A.; Bryant, S.H.; Canese, K.; Chetvernin, V.; Church, D.M.; DiCuccio, M.; Edgar, R.; Federhen, S. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2007, 36, D13–D21. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]
- Law, C.W.; Chen, Y.; Shi, W.; Smyth, G.K. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014, 15, R29. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk?/projects/fastqc/ (accessed on 21 April 2023).
- Krueger, F. Trim Galore: A Wrapper Tool around Cutadapt and FastQC to Consistently Apply Quality and Adapter Trimming to FastQ Files, with Some Extra Functionality for MspI-Digested RRBS-Type (Reduced Representation Bisufite-Seq) Libraries. 2018. Available online: http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ (accessed on 21 April 2023).
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef]
- Picard Toolkit, Broad Institute, GitHub Repository. Available online: http://broadinstitute.github.io/picard/ (accessed on 21 April 2023).
- Watanabe, K.; Taskesen, E.; Van Bochoven, A.; Posthuma, D. Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 2017, 8, 1826. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Kirkwood, T.B. A systematic look at an old problem. Nature 2008, 451, 644–647. [Google Scholar] [CrossRef] [PubMed]
- Brown-Borg, H.M. Hormonal regulation of longevity in mammals. Ageing Res. Rev. 2007, 6, 28–45. [Google Scholar] [CrossRef]
- van den Beld, A.W.; Kaufman, J.-M.; Zillikens, M.C.; Lamberts, S.W.; Egan, J.M.; van der Lely, A.J. The physiology of endocrine systems with ageing. Lancet Diabetes Endocrinol. 2018, 6, 647–658. [Google Scholar] [CrossRef]
- Bjergved, L.; Jørgensen, T.; Perrild, H.; Carlé, A.; Cerqueira, C.; Krejbjerg, A.; Laurberg, P.; Ovesen, L.; Bülow Pedersen, I.; Banke, R.L. Predictors of change in serum TSH after iodine fortification: An 11-year follow-up to the DanThyr study. J. Clin. Endocrinol. Metab. 2012, 97, 4022–4029. [Google Scholar] [CrossRef] [PubMed]
- Surks, M.I.; Ortiz, E.; Daniels, G.H.; Sawin, C.T.; Col, N.F.; Cobin, R.H.; Franklyn, J.A.; Hershman, J.M.; Burman, K.D.; Denke, M.A. Subclinical thyroid disease: Scientific review and guidelines for diagnosis and management. JAMA 2004, 291, 228–238. [Google Scholar] [CrossRef] [PubMed]
- Jansen, S.; Akintola, A.; Roelfsema, F.; Van Der Spoel, E.; Cobbaert, C.; Ballieux, B.; Egri, P.; Kvarta-Papp, Z.; Gereben, B.; Fekete, C. Human longevity is characterised by high thyroid stimulating hormone secretion without altered energy metabolism. Sci. Rep. 2015, 5, 11525. [Google Scholar] [CrossRef]
- Buffenstein, R.; Pinto, M. Endocrine function in naturally long-living small mammals. Mol. Cell. Endocrinol. 2009, 299, 101–111. [Google Scholar] [CrossRef]
- van den Beld, A.W.; Visser, T.J.; Feelders, R.A.; Grobbee, D.E.; Lamberts, S.W. Thyroid hormone concentrations, disease, physical function, and mortality in elderly men. J. Clin. Endocrinol. Metab. 2005, 90, 6403–6409. [Google Scholar] [CrossRef]
- Rozing, M.P.; Houwing-Duistermaat, J.J.; Slagboom, P.E.; Beekman, M.; Frolich, M.; de Craen, A.J.; Westendorp, R.G.; Van Heemst, D. Familial longevity is associated with decreased thyroid function. J. Clin. Endocrinol. Metab. 2010, 95, 4979–4984. [Google Scholar] [CrossRef] [PubMed]
- Chaker, L.; Cappola, A.R.; Mooijaart, S.P.; Peeters, R.P. Clinical aspects of thyroid function during ageing. Lancet Diabetes Endocrinol. 2018, 6, 733–742. [Google Scholar] [CrossRef] [PubMed]
- Morley, J.E. Hormones and the aging process. J. Am. Geriatr. Soc. 2003, 51, S333–S337. [Google Scholar] [CrossRef] [PubMed]
- Roberts, L.M.; Woodford, K.; Zhou, M.; Black, D.S.; Haggerty, J.E.; Tate, E.H.; Grindstaff, K.K.; Mengesha, W.; Raman, C.; Zerangue, N. Expression of the thyroid hormone transporters monocarboxylate transporter-8 (SLC16A2) and organic ion transporter-14 (SLCO1C1) at the blood-brain barrier. Endocrinology 2008, 149, 6251–6261. [Google Scholar] [CrossRef]
- Ciccarone, F.; Tagliatesta, S.; Caiafa, P.; Zampieri, M. DNA methylation dynamics in aging: How far are we from understanding the mechanisms? Mech. Ageing Dev. 2018, 174, 3–17. [Google Scholar] [CrossRef]
- Field, A.E.; Robertson, N.A.; Wang, T.; Havas, A.; Ideker, T.; Adams, P.D. DNA methylation clocks in aging: Categories, causes, and consequences. Mol. Cell 2018, 71, 882–895. [Google Scholar] [CrossRef]
- Miller, R.A.; Nadon, N.L. Principles of animal use for gerontological research. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2000, 55, B117–B123. [Google Scholar] [CrossRef]
- Merlo, D.F.; Rossi, L.; Pellegrino, C.; Ceppi, M.; Cardellino, U.; Capurro, C.; Ratto, A.; Sambucco, P.; Sestito, V.; Tanara, G. Cancer incidence in pet dogs: Findings of the Animal Tumor Registry of Genoa, Italy. J. Vet. Intern. Med. 2008, 22, 976–984. [Google Scholar] [CrossRef]
- Yuasa, Y. DNA methylation in cancer and ageing. Mech. Ageing Dev. 2002, 123, 1649–1654. [Google Scholar] [CrossRef]
- Fraga, M.F.; Esteller, M. Epigenetics and aging: The targets and the marks. Trends Genet. 2007, 23, 413–418. [Google Scholar] [CrossRef]
- Marques, E.; Klefström, J. Par6 family proteins in cancer. Oncoscience 2015, 2, 894. [Google Scholar] [CrossRef] [PubMed]
- Marques, E.; Englund, J.; Tervonen, T.; Virkunen, E.; Laakso, M.; Myllynen, M.; Mäkelä, A.; Ahvenainen, M.; Lepikhova, T.; Monni, O. Par6G suppresses cell proliferation and is targeted by loss-of-function mutations in multiple cancers. Oncogene 2016, 35, 1386–1398. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Wan, F.; Zhang, H.; Shi, G.; Ye, D. ITGA2B and ITGA8 are predictive of prognosis in clear cell renal cell carcinoma patients. Tumor Biol. 2016, 37, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.; Koh, Y.; Park, H.; Kim, D.Y.; Kim, D.C.; Byun, J.M.; Lee, H.J.; Yoon, S.-S. Highly expressed integrin-α8 induces epithelial to mesenchymal transition-like features in multiple myeloma with early relapse. Mol. Cells 2016, 39, 898. [Google Scholar] [CrossRef]
- Guo, Z.; Liu, J.; Su, B.; Li, X.; Zhou, L. AB179. KCNJ1 inhibits tumor proliferation and metastasis and is a prognostic factor in clear cell renal cell carcinoma. Transl. Androl. Urol. 2014, 3 (Suppl. 1), AB179. [Google Scholar]
- Pasero, C.; Gravis, G.; Granjeaud, S.; Guerin, M.; Thomassin-Piana, J.; Rocchi, P.; Salem, N.; Walz, J.; Moretta, A.; Olive, D. Highly effective NK cells are associated with good prognosis in patients with metastatic prostate cancer. Oncotarget 2015, 6, 14360. [Google Scholar] [CrossRef]
- Chen, C.-h.; Li, S.-x.; Xiang, L.-x.; Mu, H.-q.; Wang, S.-b.; Yu, K.-y. HIF-1α induces immune escape of prostate cancer by regulating NCR1/NKp46 signaling through miR-224. Biochem. Biophys. Res. Commun. 2018, 503, 228–234. [Google Scholar] [CrossRef]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef]
- Ma, Z.; Xia, W.; Liu, F.; Ma, J.; Sun, S.; Zhang, J.; Jiang, N.; Wang, X.; Hu, J.; Ma, D. SLC44A4 mutation causes autosomal dominant hereditary postlingual non-syndromic mid-frequency hearing loss. Hum. Mol. Genet. 2017, 26, 383–394. [Google Scholar] [CrossRef]
- Mattie, M.; Raitano, A.; Morrison, K.; Morrison, K.; An, Z.; Capo, L.; Verlinsky, A.; Leavitt, M.; Ou, J.; Nadell, R. The Discovery and Preclinical Development of ASG-5ME, an Antibody–Drug Conjugate Targeting SLC44A4-Positive Epithelial Tumors Including Pancreatic and Prostate CancerSLC44A4, an ADC Target for Pancreatic and Prostate Cancer. Mol. Cancer Ther. 2016, 15, 2679–2687. [Google Scholar] [CrossRef]
- Jeong, H.; Mendizabal, I.; Berto, S.; Chatterjee, P.; Layman, T.; Usui, N.; Toriumi, K.; Douglas, C.; Singh, D.; Huh, I. Evolution of DNA methylation in the human brain. Nat. Commun. 2021, 12, 2021. [Google Scholar] [CrossRef] [PubMed]
- Hannum, G.; Guinney, J.; Zhao, L.; Zhang, L.; Hughes, G.; Sadda, S.; Klotzle, B.; Bibikova, M.; Fan, J.-B.; Gao, Y. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol. Cell 2013, 49, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Goldmit, M.; Ji, Y.; Skok, J.; Roldan, E.; Jung, S.; Cedar, H.; Bergman, Y. Epigenetic ontogeny of the Igk locus during B cell development. Nat. Immunol. 2005, 6, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.R.; Busche, S.; Ge, B.; Kwan, T.; Pastinen, T.; Blanchette, M. The relationship between DNA methylation, genetic and expression inter-individual variation in untransformed human fibroblasts. Genome Biol. 2014, 15, R37. [Google Scholar] [CrossRef] [PubMed]
- Day, K.; Waite, L.L.; Thalacker-Mercer, A.; West, A.; Bamman, M.M.; Brooks, J.D.; Myers, R.M.; Absher, D. Differential DNA methylation with age displays both common and dynamic features across human tissues that are influenced by CpG landscape. Genome Biol. 2013, 14, R102. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Pathways | Term | N Gene | N Overlap | p-Value | Adj.P |
|---|---|---|---|---|---|
| GO-Biological Pathways | Hormone transport | 319 | 9 | 4.30 × 10−6 | 9.75 × 10−3 |
| Regulation of hormone levels | 513 | 11 | 5.07 × 10−6 | 9.75 × 10−3 | |
| Ion transport | 1663 | 20 | 5.87 × 10−6 | 9.75 × 10−3 | |
| Embryo development | 986 | 15 | 6.35 × 10−6 | 9.75 × 10−3 | |
| Regulation of protein modification process | 1826 | 21 | 6.63 × 10−6 | 9.75 × 10−3 | |
| Regulation of blood circulation | 293 | 8 | 1.88 × 10−5 | 2.06 × 10−2 | |
| Cardiac conduction | 143 | 6 | 1.99 × 10−5 | 2.06 × 10−2 | |
| Regulation of intracellular signal transduction | 1824 | 20 | 2.24 × 10−5 | 2.06 × 10−2 | |
| Ion transmembrane transport | 1125 | 15 | 2.96 × 10−5 | 2.42 × 10−2 | |
| Transmembrane transport | 1574 | 18 | 3.48 × 10−5 | 2.45 × 10−2 | |
| Maintenance of location | 325 | 8 | 3.93 × 10−5 | 2.45 × 10−2 | |
| Embryonic organ development | 423 | 9 | 4.01 × 10−5 | 2.45 × 10−2 | |
| Circulatory system process | 541 | 10 | 4.86 × 10−5 | 2.75 × 10−2 | |
| Regulation of myoblast differentiation | 53 | 4 | 5.27 × 10−5 | 2.77 × 10−2 | |
| Regulation of transmembrane transport | 553 | 10 | 5.84 × 10−5 | 2.86 × 10−2 | |
| Regulation of transport | 1827 | 19 | 7.43 × 10−5 | 3.40 × 10−2 | |
| Regulation of phosphorus metabolic process | 1677 | 18 | 7.86 × 10−5 | 3.40 × 10−2 | |
| Regulation of ion transmembrane transport | 467 | 9 | 8.54 × 10−5 | 3.49 × 10−2 | |
| Heterotypic cell–cell adhesion | 61 | 4 | 9.17 × 10−5 | 3.55 × 10−2 | |
| Multicellular organismal homeostasis | 477 | 9 | 1.00 × 10−4 | 3.68 × 10−2 | |
| Negative regulation of protein modification process | 599 | 10 | 1.13 × 10−4 | 3.94 × 10−2 | |
| Heart process | 287 | 7 | 1.28 × 10−4 | 4.23 × 10−2 | |
| Multicellular organismal signaling | 201 | 6 | 1.32 × 10−4 | 4.23 × 10−2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, H.S.; Jang, S.; Kim, J. Genome-Wide Integrative Transcriptional Profiling Identifies Age-Associated Signatures in Dogs. Genes 2023, 14, 1131. https://doi.org/10.3390/genes14061131
Kim HS, Jang S, Kim J. Genome-Wide Integrative Transcriptional Profiling Identifies Age-Associated Signatures in Dogs. Genes. 2023; 14(6):1131. https://doi.org/10.3390/genes14061131
Chicago/Turabian StyleKim, Hyun Seung, Subin Jang, and Jaemin Kim. 2023. "Genome-Wide Integrative Transcriptional Profiling Identifies Age-Associated Signatures in Dogs" Genes 14, no. 6: 1131. https://doi.org/10.3390/genes14061131