
Evaluating the Genetic Background Effect on Dissecting the Genetic Basis of Kernel Traits in Reciprocal Maize Introgression Lines
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Development of Reciprocal Introgression Lines
2.2. Field Experiment and Trait Measurement
2.3. DNA Extraction, SNP Genotyping, and Bin Map Construction
2.4. Data Analysis
2.5. Identification of Common QTL among Reciprocal ILs and Joint Analysis
3. Results
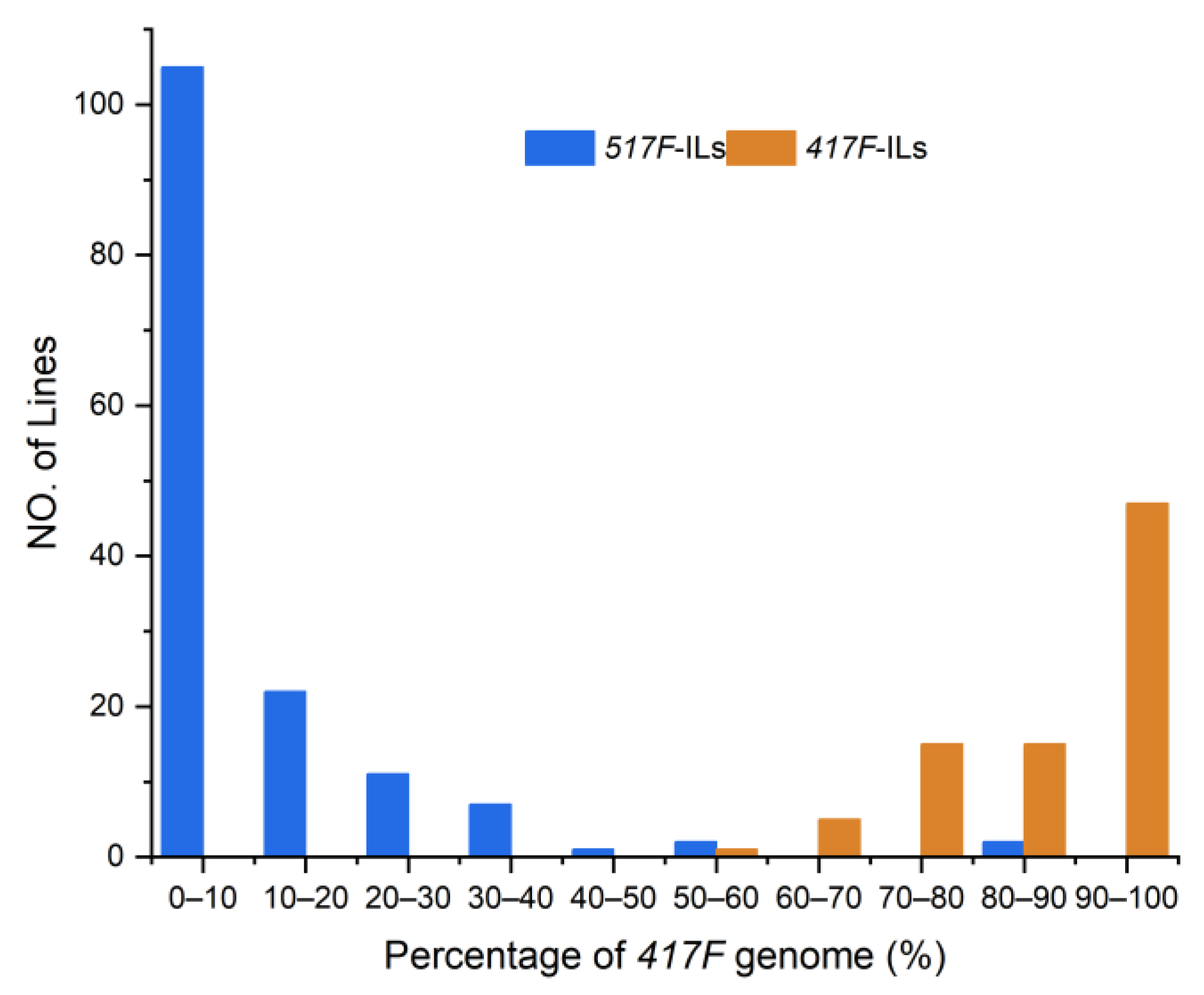
3.1. Bin Map of the Reciprocal ILs Population
3.2. Phenotypic Performances of Reciprocal ILs and Their Parents
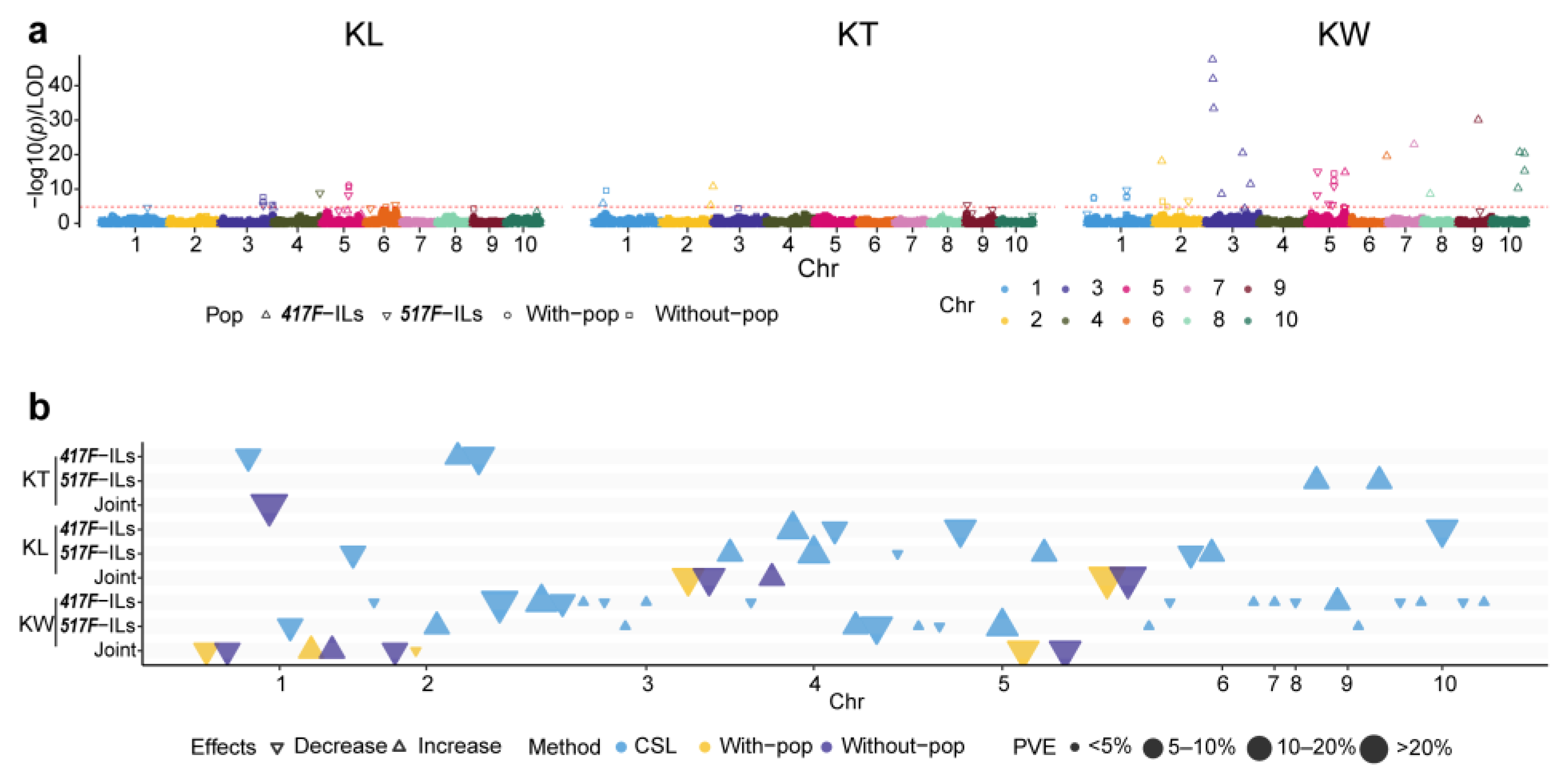
3.3. QTL Affecting Kernel Shape Traits
3.3.1. QTL of Kernel Shape Traits Identified in the Reciprocal ILs
3.3.2. QTL in Joint Analysis of the Two Reciprocal ILs for KL, KT, and KW
3.3.3. Digenic Epistatic QTL in the Reciprocal ILs for KL, KT, and KW
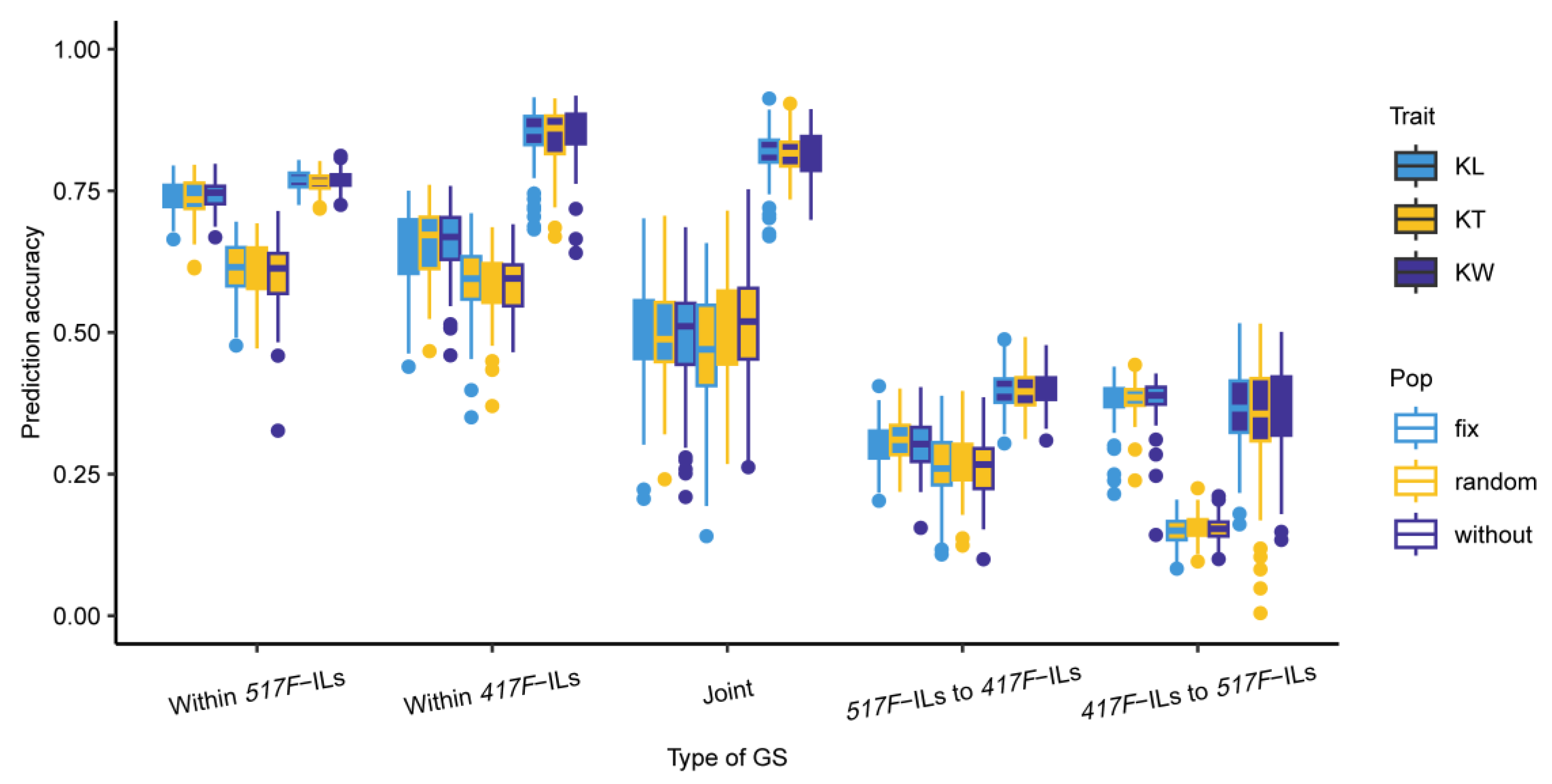
3.4. Prediction Accuracies of KL, KT, and KW Estimated with the Reciprocal ILs
4. Discussion
4.1. Genetic Background Effect on QTL of Kernel Shape Traits
4.2. Comparing the QTL Detected in this Study with Previously Reported QTL
4.3. Genetic Background Effect on Genomic Selection Accuracy
4.4. Implications in Maize Breeding
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, X.; Wang, M.; Zhang, R.; Fang, H.; Fu, X.; Yang, X.; Li, J. Genetic architecture of embryo size and related traits in maize. Crop J. 2022, 10, 204–215. [Google Scholar] [CrossRef]
- Wang, C.; Li, H.; Long, Y.; Dong, Z.; Wang, J.; Liu, C.; Wei, X.; Wan, X. A Systemic Investigation of Genetic Architecture and Gene Resources Controlling Kernel Size-Related Traits in Maize. Int. J. Mol. Sci. 2023, 24, 1025. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.K.; Rustgi, S.; Kumar, N. Genetic and molecular basis of grain size and grain number and its relevance to grain productivity in higher plants. Genome 2006, 49, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Revilla Temiño, P.; Butrón Gómez, A.M.; Malvar Pintos, R.A.; Ordás Pérez, A. Relationships among kernel weight, early vigor, and growth in maize. Crop Sci. 1999, 39, 654–658. [Google Scholar] [CrossRef]
- Dai, D.; Ma, Z.; Song, R. Maize kernel development. Mol. Breed. 2021, 41, 1–33. [Google Scholar] [CrossRef]
- Liu, M.; Tan, X.; Yang, Y.; Liu, P.; Zhang, X.; Zhang, Y.; Wang, L.; Hu, Y.; Ma, L.; Li, Z. Analysis of the genetic architecture of maize kernel size traits by combined linkage and association mapping. Plant Biotechnol. J. 2020, 18, 207–221. [Google Scholar] [CrossRef]
- Liu, J.; Huang, J.; Guo, H.; Lan, L.; Wang, H.; Xu, Y.; Yang, X.; Li, W.; Tong, H.; Xiao, Y. The conserved and unique genetic architecture of kernel size and weight in maize and rice. Plant Physiol. 2017, 175, 774–785. [Google Scholar] [CrossRef]
- Ma, J.; Cao, Y. Genetic dissection of grain yield of maize and yield-related traits through association mapping and genomic prediction. Front. Plant Sci. 2021, 12, 690059. [Google Scholar] [CrossRef]
- Qin, W.; Li, Y.-x.; Wu, X.; Li, X.; Chen, L.; Shi, Y.; Song, Y.; Zhang, D.; Wang, T.; Li, Y. Fine mapping of qKL1. 07, a major QTL for kernel length in maize. Mol. Breed. 2016, 36, 1–9. [Google Scholar] [CrossRef]
- Chen, L.; Li, Y.-X.; Li, C.; Wu, X.; Qin, W.; Li, X.; Jiao, F.; Zhang, X.; Zhang, D.; Shi, Y. Fine-mapping of qGW4. 05, a major QTL for kernel weight and size in maize. BMC Plant Biol. 2016, 16, 81. [Google Scholar] [CrossRef]
- Li, X.; Li, Y.-X.; Chen, L.; Wu, X.; Qin, W.; Song, Y.; Zhang, D.; Wang, T.; Li, Y.; Shi, Y. Fine mapping of qKW7, a major QTL for kernel weight and kernel width in maize, confirmed by the combined analytic approaches of linkage and association analysis. Euphytica 2016, 210, 221–232. [Google Scholar] [CrossRef]
- Tang, B.; Li, Y.; Mu, Z.; Chen, L.; Guo, H.; Chen, Z.; Li, C.; Liu, X.; Zhang, D.; Shi, Y. Fine mapping and candidate gene analysis of qKW7b, a major QTL for kernel width in maize. Mol. Breed. 2020, 40, 67. [Google Scholar] [CrossRef]
- Huang, J.; Lu, G.; Liu, L.; Raihan, M.S.; Xu, J.; Jian, L.; Zhao, L.; Tran, T.M.; Zhang, Q.; Liu, J. The kernel size-related quantitative trait locus qKW9 encodes a pentatricopeptide repeat protein that aaffects photosynthesis and grain filling. Plant Physiol. 2020, 183, 1696–1709. [Google Scholar] [CrossRef] [PubMed]
- Gong, D.; Tan, Z.; Zhao, H.; Pan, Z.; Sun, Q.; Qiu, F. Fine mapping of a kernel length-related gene with potential value for maize breeding. Theor. Appl. Genet. 2021, 134, 1033–1045. [Google Scholar] [CrossRef]
- Chen, H.; Zhai, L.; Chen, K.; Shen, C.; Zhu, S.; Qu, P.; Tang, J.; Liu, J.; He, H.; Xu, J. Genetic background-and environment-independent QTL and candidate gene identification of appearance quality in three MAGIC populations of rice. Front. Plant Sci. 2022, 13, 1074106. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, H.; Hu, X.; Li, K.; Liu, Z.; Wu, Y.; Huang, C. Improving genomic selection with quantitative trait loci and nonadditive effects revealed by empirical evidence in maize. Front. Plant Sci. 2019, 10, 1129. [Google Scholar] [CrossRef]
- Guo, T.; Yu, X.; Li, X.; Zhang, H.; Zhu, C.; Flint-Garcia, S.; McMullen, M.D.; Holland, J.B.; Szalma, S.J.; Wisser, R.J. Optimal designs for genomic selection in hybrid crops. Mol. Plant 2019, 12, 390–401. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, Z.; Xu, Y.; Li, P.; Zhang, X.; Xu, C. Using genomic data to improve the estimation of general combining ability based on sparse partial diallel cross designs in maize. Crop J. 2020, 8, 819–829. [Google Scholar] [CrossRef]
- Wang, X.; Pang, Y.; Zhang, J.; Zhang, Q.; Tao, Y.; Feng, B.; Zheng, T.; Xu, J.; Li, Z. Genetic background effects on QTL and QTL× environment interaction for yield and its component traits as revealed by reciprocal introgression lines in rice. Crop J. 2014, 2, 345–357. [Google Scholar] [CrossRef]
- Cheng, L.; Wang, Y.; Meng, L.; Hu, X.; Cui, Y.; Sun, Y.; Zhu, L.; Ali, J.; Xu, J.; Li, Z. Identification of salt-tolerant QTLs with strong genetic background effect using two sets of reciprocal introgression lines in rice. Genome 2012, 55, 45–55. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, H.; Li, L.; Lan, H.; Ren, Z.; Liu, D.; Wu, L.; Liu, H.; Jaqueth, J.; Li, B. Characterizing the population structure and genetic diversity of maize breeding germplasm in Southwest China using genome-wide SNP markers. BMC Genom. 2016, 17, 697. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Yang, Q.; Zheng, H.; Xu, Y.; Sang, Z.; Guo, Z.; Peng, H.; Zhang, C.; Lan, H.; Wang, Y. Genotyping by target sequencing (GBTS) and its applications. Sci. Agric. Sin. 2020, 53, 6–27. [Google Scholar]
- Chen, Z.; Wang, B.; Dong, X.; Liu, H.; Ren, L.; Chen, J.; Hauck, A.; Song, W.; Lai, J. An ultra-high density bin-map for rapid QTL mapping for tassel and ear architecture in a large F 2 maize population. BMC Genom. 2014, 15, 433. [Google Scholar] [CrossRef] [PubMed]
- Alvarado, G.; Rodríguez, F.M.; Pacheco, A.; Burgueño, J.; Crossa, J.; Vargas, M.; Pérez-Rodríguez, P.; Lopez-Cruz, M.A. META-R: A software to analyze data from multi-environment plant breeding trials. Crop J. 2020, 8, 745–756. [Google Scholar] [CrossRef]
- Ren, J.; Li, Z.; Wu, P.; Zhang, A.; Liu, Y.; Hu, G.; Cao, S.; Qu, J.; Dhliwayo, T.; Zheng, H. Genetic dissection of quantitative resistance to common rust (Puccinia sorghi) in tropical maize (Zea mays L.) by combined genome-wide association study, linkage mapping, and genomic prediction. Front. Plant Sci. 2021, 12, 692205. [Google Scholar] [CrossRef]
- Wang, J.; Wan, X.; Crossa, J.; Crouch, J.; Weng, J.; Zhai, H.; Wan, J. QTL mapping of grain length in rice (Oryza sativa L.) using chromosome segment substitution lines. Genet. Res. 2006, 88, 93–104. [Google Scholar] [CrossRef]
- Meng, L.; Li, H.; Zhang, L.; Wang, J. QTL IciMapping: Integrated software for genetic linkage map construction and quantitative trait locus mapping in biparental populations. Crop J. 2015, 3, 269–283. [Google Scholar] [CrossRef]
- Churchill, G.A.; Doerge, R. Empirical threshold values for quantitative trait mapping. Genetics 1994, 138, 963–971. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, Z. GAPIT version 3: Boosting power and accuracy for genomic association and prediction. Genom. Proteom. Bioinform. 2021, 19, 629–640. [Google Scholar] [CrossRef]
- Huang, M.; Liu, X.; Zhou, Y.; Summers, R.M.; Zhang, Z. BLINK: A package for the next level of genome-wide association studies with both individuals and markers in the millions. Gigascience 2019, 8, giy154. [Google Scholar] [CrossRef]
- Covarrubias-Pazaran, G. Genome-assisted prediction of quantitative traits using the R package sommer. PLoS ONE 2016, 11, e0156744. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.A.; van der Werf, J. Genomic best linear unbiased prediction (gBLUP) for the estimation of genomic breeding values. In Genome-Wide Association Studies and Genomic Prediction; Humana Press: Totowa, NJ, USA, 2013; pp. 321–330. [Google Scholar]
- Frascaroli, E.; Cane, M.A.; Landi, P.; Pea, G.; Gianfranceschi, L.; Villa, M.; Morgante, M.; Pe, M.E. Classical genetic and quantitative trait loci analyses of heterosis in a maize hybrid between two elite inbred lines. Genetics 2007, 176, 625–644. [Google Scholar] [CrossRef] [PubMed]
- Pan, Q.; Li, L.; Yang, X.; Tong, H.; Xu, S.; Li, Z.; Li, W.; Muehlbauer, G.J.; Li, J.; Yan, J. Genome-wide recombination dynamics are associated with phenotypic variation in maize. New Phytol. 2016, 210, 1083–1094. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Peluso, P.; Shi, J.; Liang, T.; Stitzer, M.C.; Wang, B.; Campbell, M.S.; Stein, J.C.; Wei, X.; Chin, C.-S. Improved maize reference genome with single-molecule technologies. Nature 2017, 546, 524–527. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Du, Y.; Warburton, M.L.; Xiao, Y.; Yan, J. Phenotypic plasticity contributes to maize adaptation and heterosis. Mol. Biol. Evol. 2021, 38, 1262–1275. [Google Scholar] [CrossRef]
- Zhou, Z.; Li, G.; Tan, S.; Li, D.; Weiß, T.M.; Wang, X.; Chen, S.; Würschum, T.; Liu, W. A QTL atlas for grain yield and its component traits in maize (Zea mays). Plant Breed. 2020, 139, 562–574. [Google Scholar] [CrossRef]
- Tian, F.; Bradbury, P.J.; Brown, P.J.; Hung, H.; Sun, Q.; Flint-Garcia, S.; Rocheford, T.R.; McMullen, M.D.; Holland, J.B.; Buckler, E.S. Genome-wide association study of leaf architecture in the maize nested association mapping population. Nat. Genet. 2011, 43, 159–162. [Google Scholar] [CrossRef]
- Gesteiro, N.; Cao, A.; Santiago, R.; Malvar, R.A.; Butrón, A. Genomics of maize resistance to kernel contamination with fumonisins using a multiparental advanced generation InterCross maize population (MAGIC). BMC Plant Biol. 2021, 21, 596. [Google Scholar] [CrossRef]
- Rincent, R.; Nicolas, S.; Bouchet, S.; Altmann, T.; Brunel, D.; Revilla, P.; Malvar, R.; Moreno-Gonzalez, J.; Campo, L.; Melchinger, A. Dent and Flint maize diversity panels reveal important genetic potential for increasing biomass production. Theor. Appl. Genet. 2014, 127, 2313–2331. [Google Scholar] [CrossRef]
- Li, Y.; Li, X.; Li, J.; Fu, J.; Wang, Y.; Wei, M. Dent corn genetic background influences QTL detection for grain yield and yield components in high-oil maize. Euphytica 2009, 169, 273–284. [Google Scholar] [CrossRef]
- Zhang, B.; Ma, L.; Wu, B.; Xing, Y.; Qiu, X. Introgression lines: Valuable resources for functional genomics research and breeding in rice (Oryza sativa L.). Front. Plant Sci. 2022, 13, 863789. [Google Scholar] [CrossRef] [PubMed]
- Crossa, J.; Pérez-Rodríguez, P.; Cuevas, J.; Montesinos-López, O.; Jarquín, D.; De Los Campos, G.; Burgueño, J.; González-Camacho, J.M.; Pérez-Elizalde, S.; Beyene, Y. Genomic selection in plant breeding: Methods, models, and perspectives. Trends Plant Sci. 2017, 22, 961–975. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.; Zhang, Y.; Rasheed, A.; Wang, J.; Zhang, L. Genomic prediction for grain yield and yield-related traits in chinese winter wheat. Int. J. Mol. Sci. 2020, 21, 1342. [Google Scholar] [CrossRef] [PubMed]
- Rio, S.; Mary-Huard, T.; Moreau, L.; Bauland, C.; Palaffre, C.; Madur, D.; Combes, V.; Charcosset, A. Disentangling group specific QTL allele effects from genetic background epistasis using admixed individuals in GWAS: An application to maize flowering. PLoS Genet. 2020, 16, e1008241. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Traits (mm) | Env | Parents | 517F-ILs | 417F-ILs | |||||
|---|---|---|---|---|---|---|---|---|---|
| P1 | P2 | Mean ± SD | Range | CV(%) | Mean ± SD | Range | CV(%) | ||
| KL | E1 | 10.17 | 11.10 | 9.81 ± 0.87 | 7.71–12.07 | 8.85 | 10.27 ± 0.71 | 8.21–11.77 | 6.96 |
| E2 | 8.59 | 10.03 | 9.07 ± 1.18 | 5.44–11.88 | 13.01 | 9.8 ± 0.80 | 7.91–11.46 | 8.13 | |
| E3 | 10.36 | 10.06 | 9.16 ± 1.09 | 6.04–11.32 | 11.86 | 9.40 ± 0.97 | 6.78–12.06 | 10.27 | |
| E4 | 8.08 | 10.25 | 8.46 ± 1.03 | 5.75–10.79 | 12.22 | 9.04 ± 0.93 | 6.50–11.26 | 10.31 | |
| KW | E1 | 6.76 | 9.83 | 7.30 ± 0.88 | 5.61–9.85 | 12.00 | 8.83 ± 0.74 | 6.76–10.43 | 8.39 |
| E2 | 6.66 | 8.05 | 7.22 ± 0.80 | 5.61–9.47 | 11.08 | 8.35 ± 0.70 | 6.10–10.05 | 8.37 | |
| E3 | 7.01 | 8.25 | 7.34 ± 0.75 | 5.79–9.32 | 10.29 | 8.32 ± 0.68 | 6.65–10.40 | 8.14 | |
| E4 | 7.28 | 8.81 | 7.26 ± 0.66 | 5.21–8.81 | 9.07 | 8.24 ± 0.78 | 6.34–10.36 | 9.51 | |
| KT | E1 | 4.30 | 5.78 | 4.33 ± 0.52 | 3.24–5.99 | 12.10 | 4.99 ± 0.59 | 3.71–6.84 | 11.82 |
| E2 | 3.96 | 4.96 | 4.79 ± 0.59 | 2.85–6.40 | 12.40 | 5.20 ± 0.61 | 3.37–6.75 | 11.78 | |
| E3 | 4.63 | 4.94 | 4.74 ± 0.68 | 3.64–6.85 | 14.32 | 5.17 ± 0.79 | 3.79–6.90 | 15.21 | |
| E4 | 4.95 | 5.00 | 4.75 ± 0.66 | 3.56–6.40 | 13.92 | 5.17 ± 0.79 | 3.84–7.33 | 15.22 | |
| Pop | Traits | Variance Components | h2 | |||
|---|---|---|---|---|---|---|
| 417F-ILs | KL | 0.23 *** | 0.24 *** | 0.60 *** | 0.43 | 0.54 |
| KW | 0.24 *** | 0.05 *** | 0.27 *** | 0.34 | 0.70 | |
| KT | 0.14 *** | 0.01 ** | 0.36 *** | 0.36 | 0.52 | |
| 517F-ILs | KL | 0.55 *** | 0.27 *** | 0.67 *** | 0.36 | 0.73 |
| KW | 0.27 *** | 0.002 * | 0.29 *** | 0.37 | 0.72 | |
| KT | 0.06 *** | 0.04 *** | 0.20 *** | 0.34 | 0.59 | |
| Joint | KL | 0.49 *** | 0.25 *** | 0.66 *** | 0.38 | 0.71 |
| KW | 0.54 *** | 0.01 * | 0.30 *** | 0.35 | 0.83 | |
| KT | 0.18 *** | 0.02 *** | 0.27 *** | 0.35 | 0.64 | |
| Traits | Common QTL a | M-QTL b | Chr c | Bin Marker | CSL d | Joint e | Overlap with Previous Study | ||
|---|---|---|---|---|---|---|---|---|---|
| 417F-ILs | 517F-ILs | Model with Pop | Model without Pop | ||||||
| LOD/Add/PVE (%) | LOD/Add/PVE (%) | −log10(p)/Add/PVE (%) | −log10(p)/Add/PVE (%) | ||||||
| KL | qKL1 | 1 | Bin1_225.031 | 4.57/−0.22/5.28 | KL-gCL1-3 [2] | ||||
| cqKL3a | qKL3.1 | 3 | Bin3_206.728 | 6.30/−0.15/11.95 | 7.59/−0.18/10.46 | ||||
| cqKL3a | qKL3.2 | 3 | Bin3_207.567 | 5.24/0.18/5.90 | |||||
| cqKL3b | qKL3.3 | 3 | Bin3_229.734 | 5.43/0.15/6.92 | |||||
| cqKL3b | qKL3.4 | 3 | Bin3_230.707 | 4.64/0.30/14.65 | |||||
| qKL4 | 4 | Bin4_236.121 | 8.80/0.26/10.97 | ||||||
| qKL5.1 | 5 | Bin5_8.028 | 3.13/−0.15/9.27 | ||||||
| qKL5.2 | 5 | Bin5_144.103 | 3.70/−0.21/4.15 | KL-gCL5-3 [2] | |||||
| cqKL5 | qKL5.3 | 5 | Bin5_179.183 | 3.70/−0.21/11.11 | KL-qCL5-1 [2] | ||||
| cqKL5 | qKL5.4 | 5 | Bin5_183.049 | 8.19/0.26/9.84 | KL-gCL5-4 [2], KL-qCL5-1 [2] | ||||
| cqKL5 | qKL5.5 | 5 | Bin5_184.070 | 11.06/−0.25/29.35 | 10.49/−0.24/20.65 | KL-gCL5-4 [2], KL-qCL5-1 [2] | |||
| qKL6.1 | 6 | Bin6_62.894 | 4.34/−0.22/5.01 | ||||||
| qKL6.2 | 6 | Bin6_152.563 | 5.42/0.21/5.99 | ||||||
| qKL10.2 | 10 | Bin10_142.974 | 3.37/−0.11/10.16 | KL-gCL10-1 [2] | |||||
| KT | cqKT1 | qKT1.1 | 1 | Bin1_29.052 | 5.85/−0.17/7.13 | KT-gCL1-1 [2], KT-qCL1-3 [2] | |||
| cqKT1 | qKT1.2 | 1 | Bin1_36.171 | 9.54/−0.10/50.00 | KT-qCL1-5 [2] S1_35756298 [36] | ||||
| cqKT2 | qKT2.1 | 2 | Bin2_231.109 | 5.22/0.14/6.32 | |||||
| cqKT2 | qKT2.2 | 2 | Bin2_236.423 | 10.74/−0.23/15.32 | |||||
| qKT9.1 | 9 | Bin9_7.716 | 5.30/0.11/8.88 | ||||||
| qKT9.2 | 9 | Bin9_147.413 | 3.97/0.09/6.64 | KT-qCL9-2 [2] | |||||
| KW | qKW1.1 | 1 | Bin1_22.486 | 7.57/−0.19/9.47 | 7.32/−0.19/8.80 | ||||
| qKW1.2 | 1 | Bin1_206.512 | 9.70/−0.23/8.14 | 7.94/0.17/7.59 | 7.51/0.17/5.05 | KW-qCL1-4 [2] | |||
| cqKW2 | qKW2.1 | 2 | Bin2_14.332 | 18.10/−0.25/2.78 | MQTL_GW_8 [37] | ||||
| cqKW2 | qKW2.2 | 2 | Bin2_15.379 | 6.46/−0.17/5.90 | |||||
| cqKW2 | qKW2.3 | 2 | Bin2_31.026 | 4.86/−0.14/4.12 | |||||
| qKW2.4 | 2 | Bin2_190.258 | 6.51/0.19/5.60 | MQTL_GW_11 [37] | |||||
| cqKW3a | qKW3.1 | 3 | Bin3_12.511 | 47.62/−0.63/21.29 | |||||
| cqKW3a | qKW3.2 | 3 | Bin3_13.777 | 41.97/0.56/15.60 | KW-qCL3-5 [2] | ||||
| cqKW3a | qKW3.3 | 3 | Bin3_15.625 | 33.45/−0.41/9.09 | |||||
| qKW3.4 | 3 | Bin3_56.351 | 8.56/0.14/1.03 | ||||||
| cqKW3b | qKW3.5 | 3 | Bin3_181.586 | 20.50/−0.23/3.50 | KW-qCL3-8 [2] | ||||
| cqKW3b | qKW3.6 | 3 | Bin3_188.137 | 3.63/0.12/2.84 | |||||
| cqKW3b | qKW3.7 | 3 | Bin3_189.058 | 4.30/0.09/0.45 | |||||
| qKW3.8 | 3 | Bin3_210.382 | 11.44/−0.15/1.48 | KW-gCL3-2 [2] | |||||
| cqKW5a | qKW5.1 | 5 | Bin5_30.612 | 8.27/0.30/6.84 | MQTL_GW_25 [37] | ||||
| cqKW5a | qKW5.2 | 5 | Bin5_36.708 | 14.98/−0.36/13.72 | KW-gCL5-2 [2] | ||||
| cqKW5b | qKW5.3 | 5 | Bin5_167.222 | 5.64/0.17/4.7 | |||||
| cqKW5b | qKW5.4 | 5 | Bin5_175.956 | 5.40/−0.19/4.96 | |||||
| cqKW5b | qKW5.5 | 5 | Bin5_182.560 | 10.81/0.25/10.48 | 12.43/−0.27/15.77 | ||||
| cqKW5b | qKW5.6 | 5 | Bin5_183.049 | 14.43/−0.29/16.05 | |||||
| cqKW5c | qKW5.7 | 5 | Bin5_208.649 | 4.50/0.15/3.78 | KW-qCL5-4 [2] | ||||
| cqKW5c | qKW5.8 | 5 | Bin5_208.770 | 14.86/−0.31/2.07 | KW-qCL5-4 [2] | ||||
| qKW6 | 6 | Bin6_165.776 | 19.54/0.26/3.33 | ||||||
| qKW7 | 7 | Bin7_144.792 | 22.95/0.37/4.16 | KW-qCL7-5 [2] | |||||
| qKW8 | 8 | Bin8_20.414 | 8.58/−0.13/1.06 | ||||||
| cqKW9 | qKW9.1 | 9 | Bin9_124.092 | 30.09/0.38/7.10 | |||||
| cqKW9 | qKW9.2 | 9 | Bin9_133.648 | 3.50/0.14/2.78 | MQTL_GW_40 [37] | ||||
| cqKW10 | qKW10.1 | 10 | Bin10_137.330 | 10.26/−0.14/1.32 | KW-gCL10-3 [2] | ||||
| cqKW10 | qKW10.2 | 10 | Bin10_139.438 | 20.70/0.21/3.69 | KW-gCL10-3 [2] | ||||
| cqKW10 | qKW10.3 | 10 | Bin10_145.998 | 20.38/−0.26/3.76 | KW-gCL10-3 [2] | ||||
| cqKW10 | qKW10.4 | 10 | Bin10_146.294 | 15.26/0.21/2.30 | KW-gCL10-3 [2], KW-qCL10-1 [2] | ||||
| Traits | Bin Marker 1 | Bin Marker 2 | LOD Aa a | LOD Total b | PVE aa (%) c | PVE Total (%) d | Add1 e | Add2 f | Add by Add g | Pop |
|---|---|---|---|---|---|---|---|---|---|---|
| KL | Bin3_196.231 | Bin3_207.567 | 7.46 | 7.75 | 10.11 | 15.98 | 0.02 | 0.22 | 0.1545 | 517F-ILs |
| KW | Bin1_206.512 | Bin1_215.108 | 7.01 | 11.19 | 3.89 | 15.59 | −0.34 | 0.09 | −0.1232 | 517F-ILs |
| KW | Bin5_181.969 | Bin5_182.560 | 7.78 | 11.53 | 3.23 | 18.41 | −0.09 | 0.33 | −0.1392 | 517F-ILs |
| KW | Bin3_56.351 | Bin7_34.740 | 8.52 | 10.53 | 0.29 | 1.27 | 0.10 | −0.02 | −0.0885 | 417F-ILs |
| KW | Bin9_18.667 | Bin10_137.330 | 7.56 | 12.17 | 0.22 | 1.57 | −0.02 | −0.20 | −0.0827 | 417F-ILs |
| KW | Bin9_18.667 | Bin10_139.438 | 7.50 | 22.61 | 0.19 | 4.08 | −0.02 | 0.15 | −0.0819 | 417F-ILs |
| KW | Bin9_21.715 | Bin10_137.330 | 7.38 | 11.88 | 0.22 | 1.55 | −0.02 | −0.20 | −0.0822 | 417F-ILs |
| KW | Bin9_21.715 | Bin10_139.438 | 7.31 | 22.32 | 0.18 | 4.06 | −0.02 | 0.15 | −0.0813 | 417F-ILs |
| KW | Bin9_99.947 | Bin9_124.092 | 7.92 | 30.70 | 0.44 | 7.22 | −0.09 | 0.47 | 0.0847 | 417F-ILs |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, R.; Cui, Y.; Kong, L.; Zheng, F.; Zhao, W.; Meng, Q.; Yuan, J.; Zhang, M.; Chen, Y. Evaluating the Genetic Background Effect on Dissecting the Genetic Basis of Kernel Traits in Reciprocal Maize Introgression Lines. Genes 2023, 14, 1044. https://doi.org/10.3390/genes14051044
Liu R, Cui Y, Kong L, Zheng F, Zhao W, Meng Q, Yuan J, Zhang M, Chen Y. Evaluating the Genetic Background Effect on Dissecting the Genetic Basis of Kernel Traits in Reciprocal Maize Introgression Lines. Genes. 2023; 14(5):1044. https://doi.org/10.3390/genes14051044
Chicago/Turabian StyleLiu, Ruixiang, Yakun Cui, Lingjie Kong, Fei Zheng, Wenming Zhao, Qingchang Meng, Jianhua Yuan, Meijing Zhang, and Yanping Chen. 2023. "Evaluating the Genetic Background Effect on Dissecting the Genetic Basis of Kernel Traits in Reciprocal Maize Introgression Lines" Genes 14, no. 5: 1044. https://doi.org/10.3390/genes14051044