
A Novel 13q12 Microdeletion Associated with Familial Syndromic Corneal Opacification

Abstract
:1. Introduction
2. Materials and Methods
2.1. Clinical Testing
2.2. RNAseq Analysis
3. Results
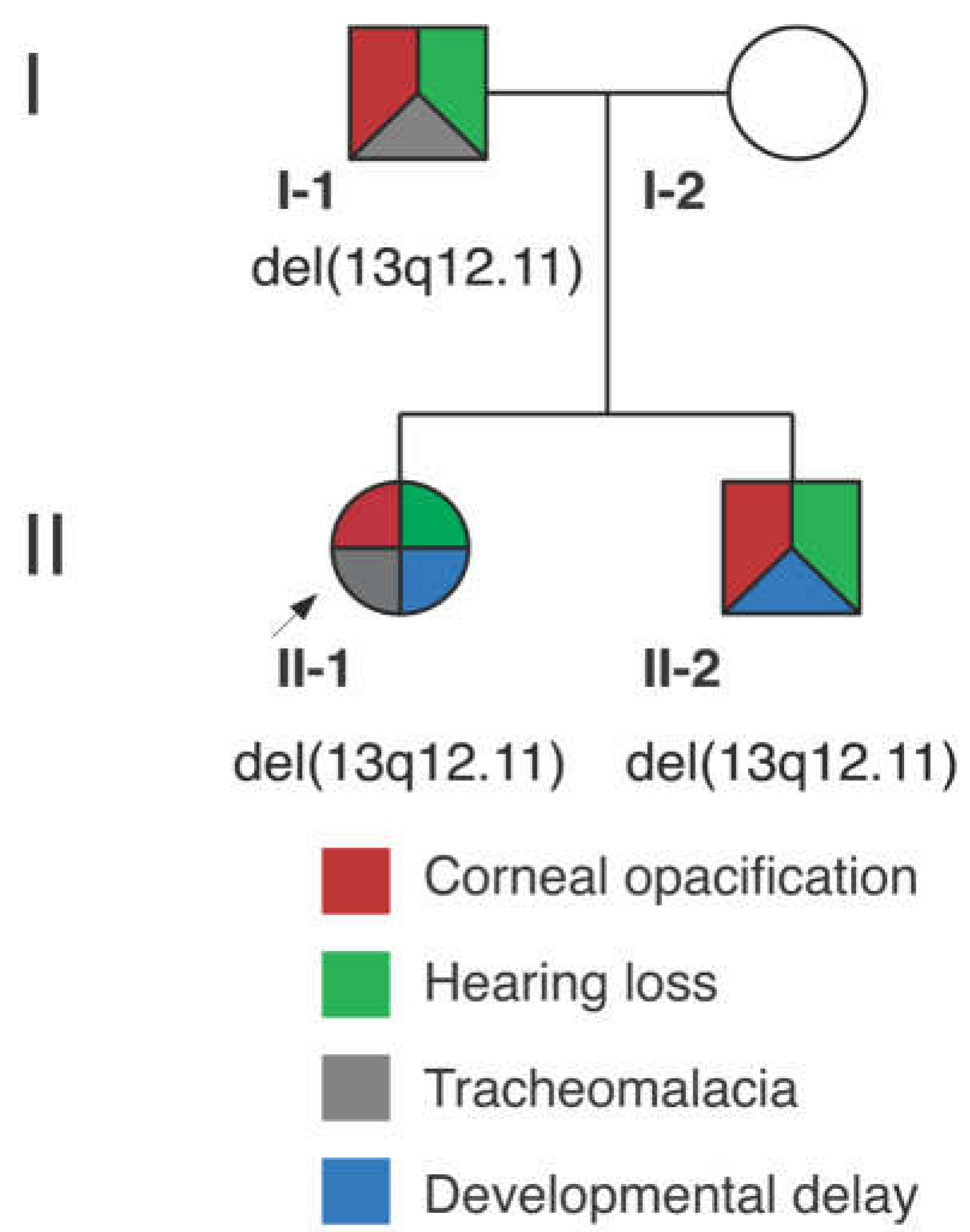
3.1. Clinical Report
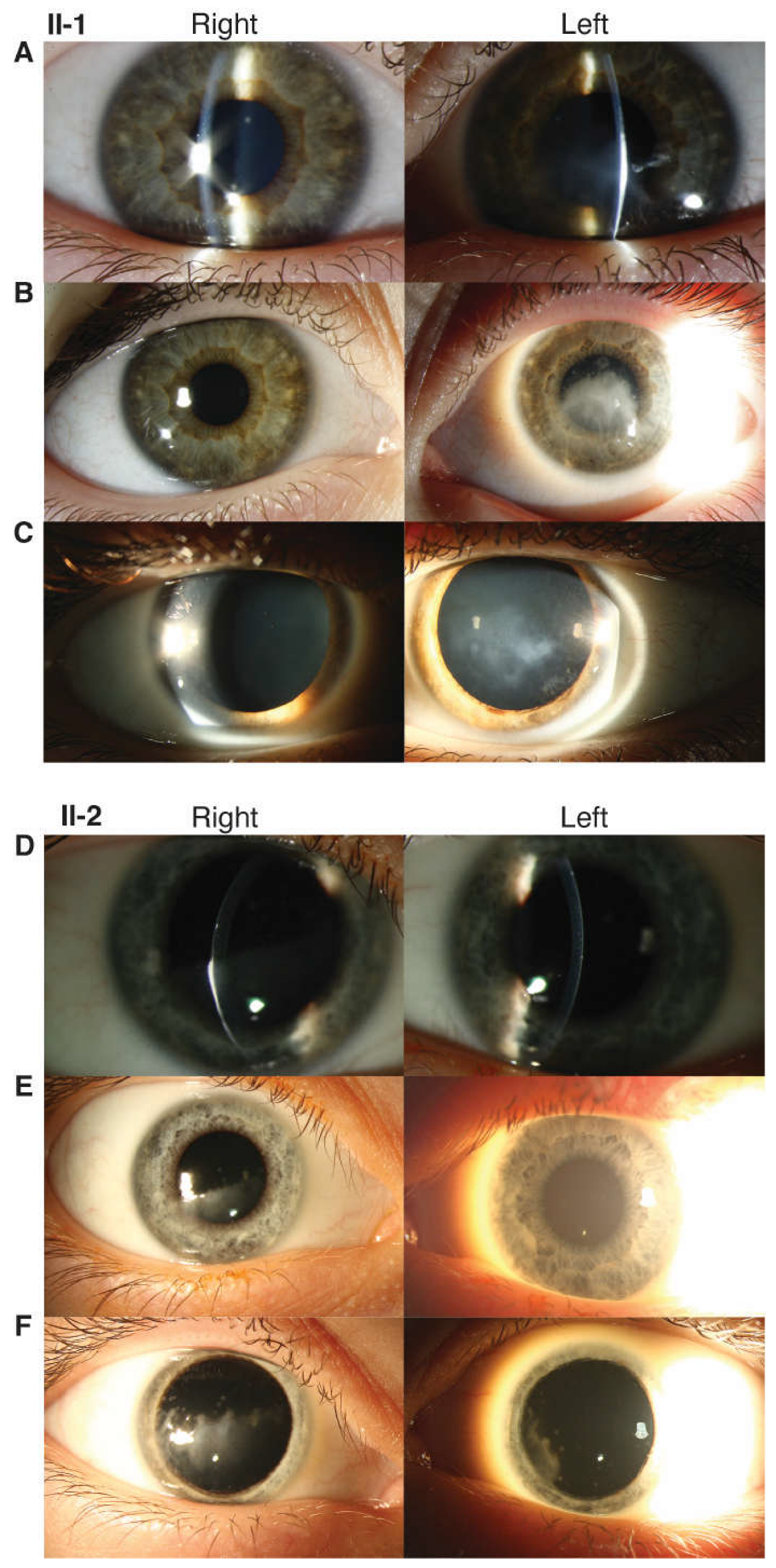
3.1.1. Proband (II-1)
3.1.2. Proband’s Brother (II-2)
3.1.3. Proband’s Parents
3.2. Genetic Analysis
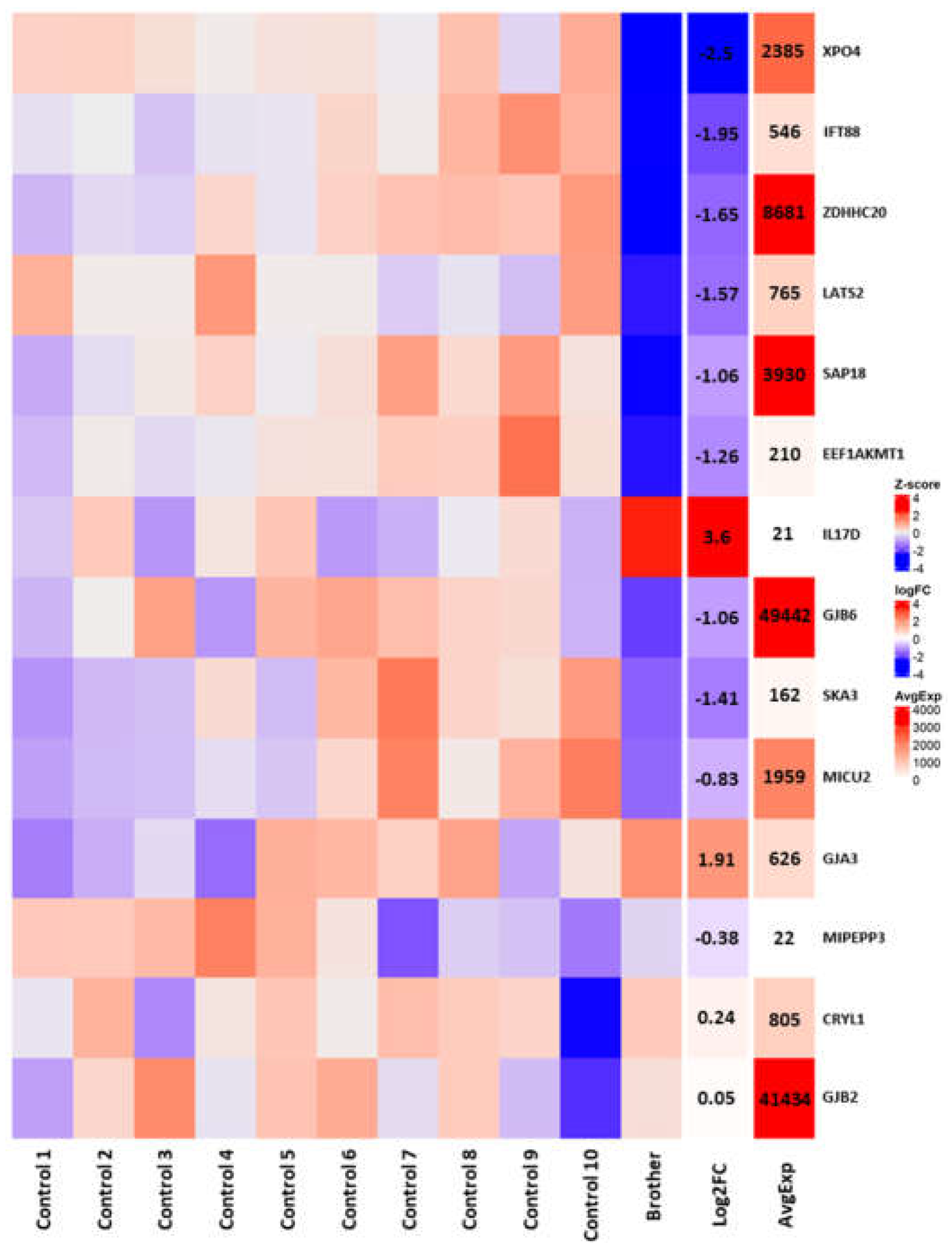
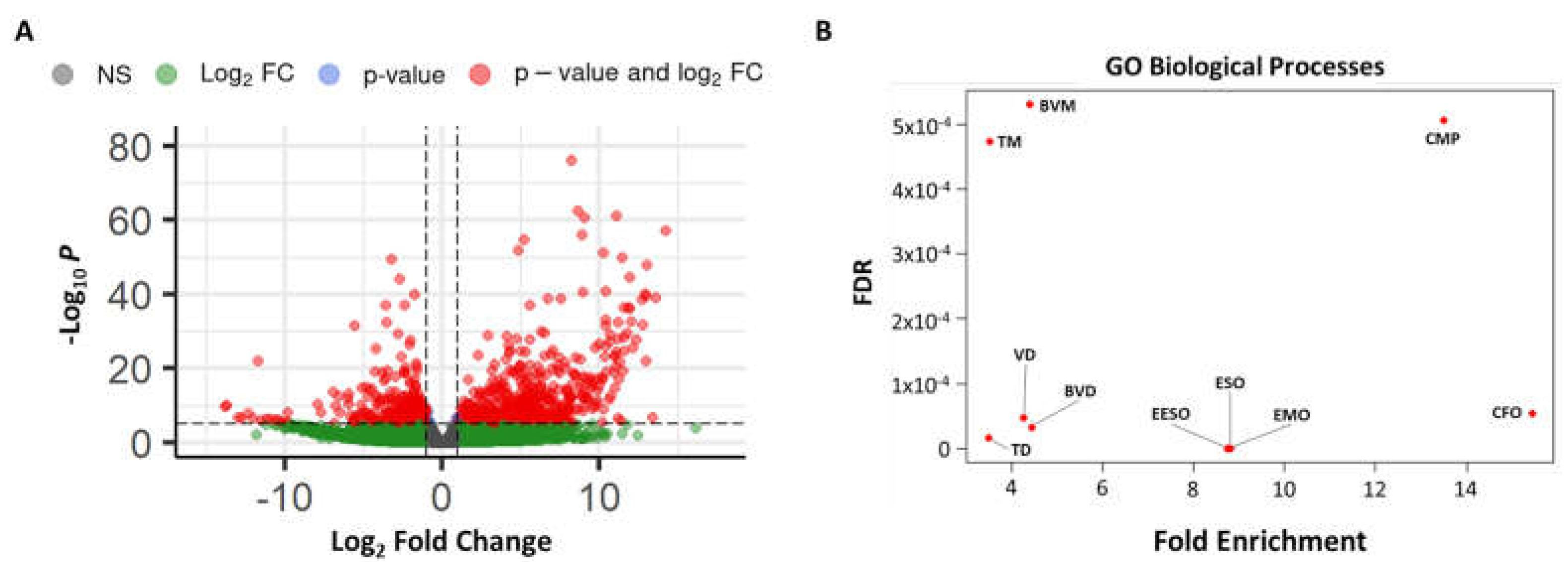
3.3. RNAseq Analysis
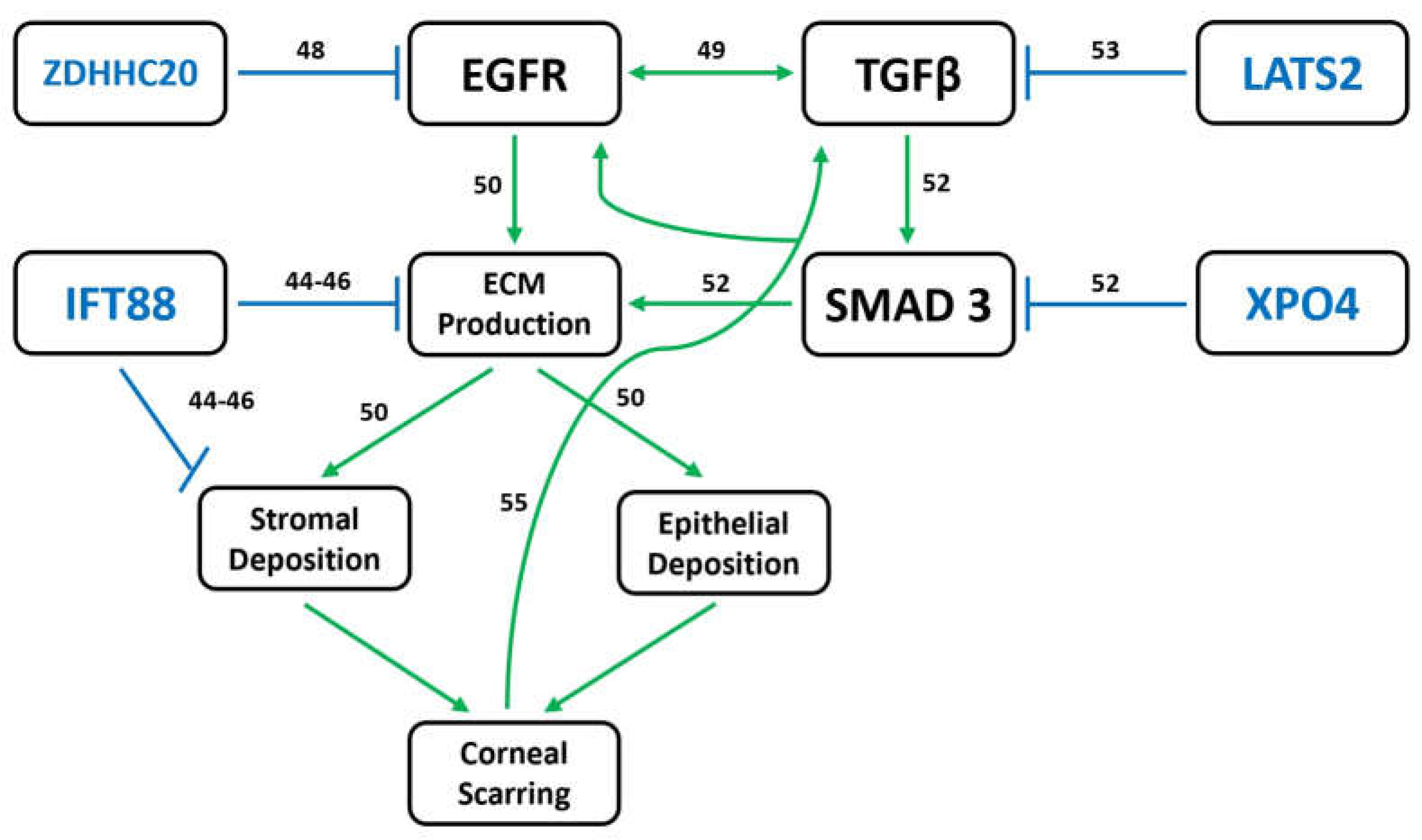
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bourges, J.L. Corneal dystrophies. J. Français d’Ophtalmol. 2017, 40, e177–e192. [Google Scholar] [CrossRef] [PubMed]
- Klintworth, G.K. Corneal dystrophies. Orphanet J. Rare Dis. 2009, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- Weiss, J.S.; Moller, H.U.; Lisch, W.; Kinoshita, S.; Aldave, A.J.; Belin, M.W.; Kivela, T.; Busin, M.; Munier, F.L.; Seitz, B.; et al. The IC3D classification of the corneal dystrophies. Cornea 2008, 27 (Suppl. S2), S1–S83. [Google Scholar] [CrossRef] [PubMed]
- Kannabiran, C.; Chaurasia, S.; Ramappa, M.; Mootha, V.V. Update on the genetics of corneal endothelial dystrophies. Indian. J. Ophthalmol. 2022, 70, 2239–2248. [Google Scholar] [CrossRef]
- Lisch, W.; Weiss, J.S. Clinical and genetic update of corneal dystrophies. Exp. Eye Res. 2019, 186, 107715. [Google Scholar] [CrossRef]
- Kannabiran, C.; Klintworth, G.K. TGFBI gene mutations in corneal dystrophies. Hum. Mutat. 2006, 27, 615–625. [Google Scholar] [CrossRef]
- Han, K.E.; Choi, S.I.; Kim, T.I.; Maeng, Y.S.; Stulting, R.D.; Ji, Y.W.; Kim, E.K. Pathogenesis and treatments of TGFBI corneal dystrophies. Prog. Retin. Eye Res. 2016, 50, 67–88. [Google Scholar] [CrossRef]
- Black, G.C.M.; Ashworth, J.L.; Sergouniotis, P.I. (Eds.) Genetic disorders affecting the cornea. In Clinical Ophthalmic Genetics and Genomics; Academic Press: Cambridge, MA, USA, 2022. [Google Scholar] [CrossRef]
- Skeens, H.M.; Brooks, B.P.; Holland, E.J. Congenital aniridia variant: Minimally abnormal irides with severe limbal stem cell deficiency. Ophthalmology 2011, 118, 1260–1264. [Google Scholar] [CrossRef]
- Strungaru, M.H.; Mah, D.; Chan, C.C. Focal limbal stem cell deficiency in Turner syndrome: Report of two patients and review of the literature. Cornea 2014, 33, 207–209. [Google Scholar] [CrossRef]
- Fernandes, M.; Sangwan, V.S.; Vemuganti, G.K. Limbal stem cell deficiency and xeroderma pigmentosum: A case report. Eye 2004, 18, 741–743. [Google Scholar] [CrossRef]
- Messmer, E.M.; Kenyon, K.R.; Rittinger, O.; Janecke, A.R.; Kampik, A. Ocular manifestations of keratitis-ichthyosis-deafness (KID) syndrome. Ophthalmology 2005, 112, e1–e6. [Google Scholar] [CrossRef] [PubMed]
- Der Kaloustian, V.M.; Russell, L.; Aradhya, S.; Richard, G.; Rosenblatt, B.; Melancon, S. A de novo 2.1-Mb deletion of 13q12.11 in a child with developmental delay and minor dysmorphic features. Am. J. Med. Genet. Part A 2011, 155A, 2538–2542. [Google Scholar] [CrossRef] [PubMed]
- Lagou, M.; Papoulidis, I.; Orru, S.; Papadopoulos, V.; Daskalakis, G.; Kontodiou, M.; Anastasakis, E.; Petersen, M.B.; Kitsos, G.; Thomaidis, L.; et al. A de novo 2.9 Mb interstitial deletion at 13q12.11 in a child with developmental delay accompanied by mild dysmorphic characteristics. Mol. Cytogenet. 2014, 7, 92. [Google Scholar] [CrossRef]
- Tanteles, G.A.; Dixit, A.; Smith, N.; Martin, K.; Suri, M. Mild phenotype in a patient with a de-novo 2.9-Mb interstitial deletion at 13q12.11. Clin. Dysmorphol. 2011, 20, 61–65. [Google Scholar] [CrossRef]
- Firth, H.V.; Richards, S.M.; Bevan, A.P.; Clayton, S.; Corpas, M.; Rajan, D.; Van Vooren, S.; Moreau, Y.; Pettett, R.M.; Carter, N.P. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans using Ensembl Resources. Am. J. Hum. Genet. 2009, 84, 524–533. [Google Scholar] [CrossRef] [PubMed]
- Agilent 2200 TapeStation User Manual (p/n G2964-90002). 2016. Available online: https://www.agilent.com/cs/library/usermanuals/public/G2964-90000_TapeStation_USR_ENU.pdf (accessed on 12 February 2023).
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A quality control tool for high throughput sequence data. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 12 February 2023).
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- You, J.; Corley, S.M.; Wen, L.; Hodge, C.; Hollhumer, R.; Madigan, M.C.; Wilkins, M.R.; Sutton, G. RNA-Seq analysis and comparison of corneal epithelium in keratoconus and myopia patients. Sci. Rep. 2018, 8, 389. [Google Scholar] [CrossRef]
- Mi, H.; Muruganujan, A.; Ebert, D.; Huang, X.; Thomas, P.D. PANTHER version 14: More genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 2019, 47, D419–D426. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Blighe, K.; Rana, S.; Lewis, M. EnhancedVolcano: Publication-ready volcano plots with enhanced colouring and labeling. 2018. Available online: https://bioconductor.org/packages/devel/bioc/vignettes/EnhancedVolcano/inst/doc/EnhancedVolcano.htm (accessed on 25 April 2023).
- Downie, L.; Halliday, J.; Burt, R.; Lunke, S.; Lynch, E.; Martyn, M.; Poulakis, Z.; Gaff, C.; Sung, V.; Wake, M.; et al. Exome sequencing in infants with congenital hearing impairment: A population-based cohort study. Eur. J. Hum. Genet. 2020, 28, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.E.; Mohan, R.R.; Mohan, R.R.; Ambrosio, R., Jr.; Hong, J.; Lee, J. The corneal wound healing response: Cytokine-mediated interaction of the epithelium, stroma, and inflammatory cells. Prog. Retin. Eye Res. 2001, 20, 625–637. [Google Scholar] [CrossRef]
- Srinivas, M.; Verselis, V.K.; White, T.W. Human diseases associated with connexin mutations. Biochim. Biophys. Acta (BBA)-Biomembr. 2018, 1860, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Pavone, P.; Briuglia, S.; Falsaperla, R.; Warm, A.; Pavone, V.; Bernardini, L.; Novelli, A.; Pratico, A.D.; Salpietro, V.; Ruggieri, M. Wide spectrum of congenital anomalies including choanal atresia, malformed extremities, and brain and spinal malformations in a girl with a de novo 5.6-Mb deletion of 13q12.11-13q12.13. Am. J. Med. Genet. Part A 2014, 164A, 1734–1743. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.M.; Baross, A.; Delaney, A.D.; Ally, A.; Arbour, L.; Armstrong, L.; Asano, J.; Bailey, D.K.; Barber, S.; Birch, P.; et al. Oligonucleotide microarray analysis of genomic imbalance in children with mental retardation. Am. J. Hum. Genet. 2006, 79, 500–513. [Google Scholar] [CrossRef] [PubMed]
- Gerido, D.A.; White, T.W. Connexin disorders of the ear, skin, and lens. Biochim. Biophys. Acta (BBA)-Biomembr. 2004, 1662, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Online Mendelian Inheritance in Man, OMIM. McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University (Baltimore, MD), {2023}. Available online: https://omim.org/ (accessed on 12 February 2023).
- Kasmann, B.; Ruprecht, K.W. Ocular manifestations in a father and son with EEC syndrome. Graefes Arch. Clin. Exp. Ophthalmol. 1997, 235, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Felipe, A.F.; Abazari, A.; Hammersmith, K.M.; Rapuano, C.J.; Nagra, P.K.; Peiro, B.M. Corneal changes in ectrodactyly-ectodermal dysplasia-cleft lip and palate syndrome: Case series and literature review. Int. Ophthalmol. 2012, 32, 475–480. [Google Scholar] [CrossRef]
- Tyagi, P.; Tyagi, V.; Hashim, A.A. Ocular and non-ocular manifestations of hypohidrotic ectodermal dysplasia. BMJ Case Rep. 2011, 2011, bcr0120113731. [Google Scholar] [CrossRef]
- Landau Prat, D.; Katowitz, W.R.; Strong, A.; Katowitz, J.A. Ocular manifestations of ectodermal dysplasia. Orphanet J. Rare Dis. 2021, 16, 197. [Google Scholar] [CrossRef] [PubMed]
- Hoefsloot, L.H.; Roux, A.F.; Bitner-Glindzicz, M. EMQN Best Practice guidelines for diagnostic testing of mutations causing non-syndromic hearing impairment at the DFNB1 locus. Eur. J. Hum. Genet. 2013, 21, 1325–1329. [Google Scholar] [CrossRef] [PubMed]
- Del Castillo, F.J.; Del Castillo, I. DFNB1 Non-syndromic Hearing Impairment: Diversity of Mutations and Associated Phenotypes. Front. Mol. Neurosci. 2017, 10, 428. [Google Scholar] [CrossRef] [PubMed]
- del Castillo, I.; Villamar, M.; Moreno-Pelayo, M.A.; del Castillo, F.J.; Alvarez, A.; Telleria, D.; Menendez, I.; Moreno, F. A deletion involving the connexin 30 gene in nonsyndromic hearing impairment. N. Engl. J. Med. 2002, 346, 243–249. [Google Scholar] [CrossRef]
- Sobreira, N.; Schiettecatte, F.; Valle, D.; Hamosh, A. GeneMatcher: A matching tool for connecting investigators with an interest in the same gene. Hum. Mutat. 2015, 36, 928–930. [Google Scholar] [CrossRef]
- International Mouse Phenotyping Consortium. Available online: https://www.mousephenotype.org/data/genes/MGI:1888526 (accessed on 1 December 2022).
- Portal, C.; Rompolas, P.; Lwigale, P.; Iomini, C. Primary cilia deficiency in neural crest cells models anterior segment dysgenesis in mouse. eLife 2019, 8, e52423. [Google Scholar] [CrossRef]
- Collins, I.; Wann, A.K.T. Regulation of the Extracellular Matrix by Ciliary Machinery. Cells 2020, 9, 278. [Google Scholar] [CrossRef]
- Toomer, K.A.; Fulmer, D.; Guo, L.; Drohan, A.; Peterson, N.; Swanson, P.; Brooks, B.; Mukherjee, R.; Body, S.; Lipschutz, J.H.; et al. A role for primary cilia in aortic valve development and disease. Dev. Dyn. 2017, 246, 625–634. [Google Scholar] [CrossRef]
- Coveney, C.R.; Collins, I.; Mc Fie, M.; Chanalaris, A.; Yamamoto, K.; Wann, A.K.T. Cilia protein IFT88 regulates extracellular protease activity by optimizing LRP-1-mediated endocytosis. FASEB J. 2018, 32, fj201800334. [Google Scholar] [CrossRef]
- Zaki, M.S.; Sattar, S.; Massoudi, R.A.; Gleeson, J.G. Co-occurrence of distinct ciliopathy diseases in single families suggests genetic modifiers. Am. J. Med. Genet. Part A 2011, 155A, 3042–3049. [Google Scholar] [CrossRef]
- Kadry, Y.A.; Lee, J.Y.; Witze, E.S. Regulation of EGFR signalling by palmitoylation and its role in tumorigenesis. Open. Biol. 2021, 11, 210033. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Bazan, H.E. Epidermal growth factor synergism with TGF-beta1 via PI-3 kinase activity in corneal keratocyte differentiation. Investig. Ophthalmol. Vis. Sci. 2008, 49, 2936–2945. [Google Scholar] [CrossRef] [PubMed]
- Shu, D.Y.; Hutcheon, A.E.K.; Zieske, J.D.; Guo, X. Epidermal Growth Factor Stimulates Transforming Growth Factor-Beta Receptor Type II Expression In Corneal Epithelial Cells. Sci. Rep. 2019, 9, 8079. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alfoldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Metwally, M.; Bayoumi, A.; Khan, A.; Adams, L.A.; Aller, R.; Garcia-Monzon, C.; Arias-Loste, M.T.; Bugianesi, E.; Miele, L.; Anna, A.; et al. Copy number variation and expression of exportin-4 associates with severity of fibrosis in metabolic associated fatty liver disease. eBioMedicine 2021, 70, 103521. [Google Scholar] [CrossRef]
- Piersma, B.; Bank, R.A.; Boersema, M. Signaling in Fibrosis: TGF-beta, WNT, and YAP/TAZ Converge. Front. Med. 2015, 2, 59. [Google Scholar] [CrossRef] [PubMed]
- Souzeau, E.; Siggs, O.M.; Mullany, S.; Schmidt, J.M.; Hassall, M.M.; Dubowsky, A.; Chappell, A.; Breen, J.; Bae, H.; Nicholl, J.; et al. Diagnostic yield of candidate genes in an Australian corneal dystrophy cohort. Mol. Genet. Genomic. Med. 2022, 10, e2023. [Google Scholar] [CrossRef] [PubMed]
- Parker, M.W.; Rossi, D.; Peterson, M.; Smith, K.; Sikstrom, K.; White, E.S.; Connett, J.E.; Henke, C.A.; Larsson, O.; Bitterman, P.B. Fibrotic extracellular matrix activates a profibrotic positive feedback loop. J. Clin. Investig. 2014, 124, 1622–1635. [Google Scholar] [CrossRef]
- Amador, C.; Shah, R.; Ghiam, S.; Kramerov, A.A.; Ljubimov, A.V. Gene Therapy in the Anterior Eye Segment. Curr. Gene Ther. 2022, 22, 104–131. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variant (hg19 Coordinates) | Overlapping Phenotype | Includes XPO4/CRYL1 | Reference |
|---|---|---|---|
| del 13: 20,079,051–25,514,640 | Laryngomalacia; motor, language, and speech delays | XPO4, CRYL1 | DECIPHER ID 282712 |
| del 13: 20,407,295–22,453,812 | Developmental delay | XPO4, CRYL1 | DECIPHER ID 317099 |
| del 13: 19,938,561–22,840,254 | Developmental delay, speech delay | XPO4, CRYL1 | Lagou et al. [14] |
| del 13: 20,174,448–23,128,904 | Developmental delay affecting speech and language, recurrent otitis media, conductive hearing loss | XPO4, CRYL1 | Tanteles et al. [15] |
| del 13: 20,521,989–22,617,211 | Developmental delay | XPO4, CRYL1 | Der Kaloustian et al. [13] |
| del 13: 20,808,367–21,001,431 | Intellectual disability, sensorineural hearing impairment | CRYL1 * | DECIPHER ID 273408 |
| del 13: 20,808,544–21,078,913 | Bilateral conductive hearing impairment | CRYL1 * | DECIPHER ID 379530 |
| del 13: 20,797,139–21,059,969 | Intellectual disability, sensorineural hearing impairment | CRYL1 | DECIPHER ID 285395 |
| del 13: 20,281,273–21,945,915 | Laryngomalacia, stridor | XPO4, CRYL1 | DECIPHER ID 384469 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Serpen, J.Y.; Presley, W.; Beil, A.; Armenti, S.T.; Johnson, K.; Mian, S.I.; Innis, J.W.; Prasov, L. A Novel 13q12 Microdeletion Associated with Familial Syndromic Corneal Opacification. Genes 2023, 14, 1034. https://doi.org/10.3390/genes14051034
Serpen JY, Presley W, Beil A, Armenti ST, Johnson K, Mian SI, Innis JW, Prasov L. A Novel 13q12 Microdeletion Associated with Familial Syndromic Corneal Opacification. Genes. 2023; 14(5):1034. https://doi.org/10.3390/genes14051034
Chicago/Turabian StyleSerpen, Jasmine Y., William Presley, Adelyn Beil, Stephen T. Armenti, Kayla Johnson, Shahzad I. Mian, Jeffrey W. Innis, and Lev Prasov. 2023. "A Novel 13q12 Microdeletion Associated with Familial Syndromic Corneal Opacification" Genes 14, no. 5: 1034. https://doi.org/10.3390/genes14051034