
Epidemiology of PAX6 Gene Pathogenic Variants and Expected Prevalence of PAX6-Associated Congenital Aniridia across the Russian Federation: A Nationwide Study
 , , , , , ,
, , , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples
2.1.1. The Study Sample
2.1.2. Epidemiology Study Data
2.1.3. Official Statistical Population Data
2.2. DNA Isolation
2.3. Molecular Genetic Diagnostics
2.4. Statistical Analysis
3. Results
3.1. Clinical Characteristics of a New Sample Comprising 100 Aniridia Families, Totaling 129 Patients
3.2. New Data on PAX6 Gene Variants Have Been Identified in a Sample Comprising 100 Aniridia Families, Totaling 129 Patients
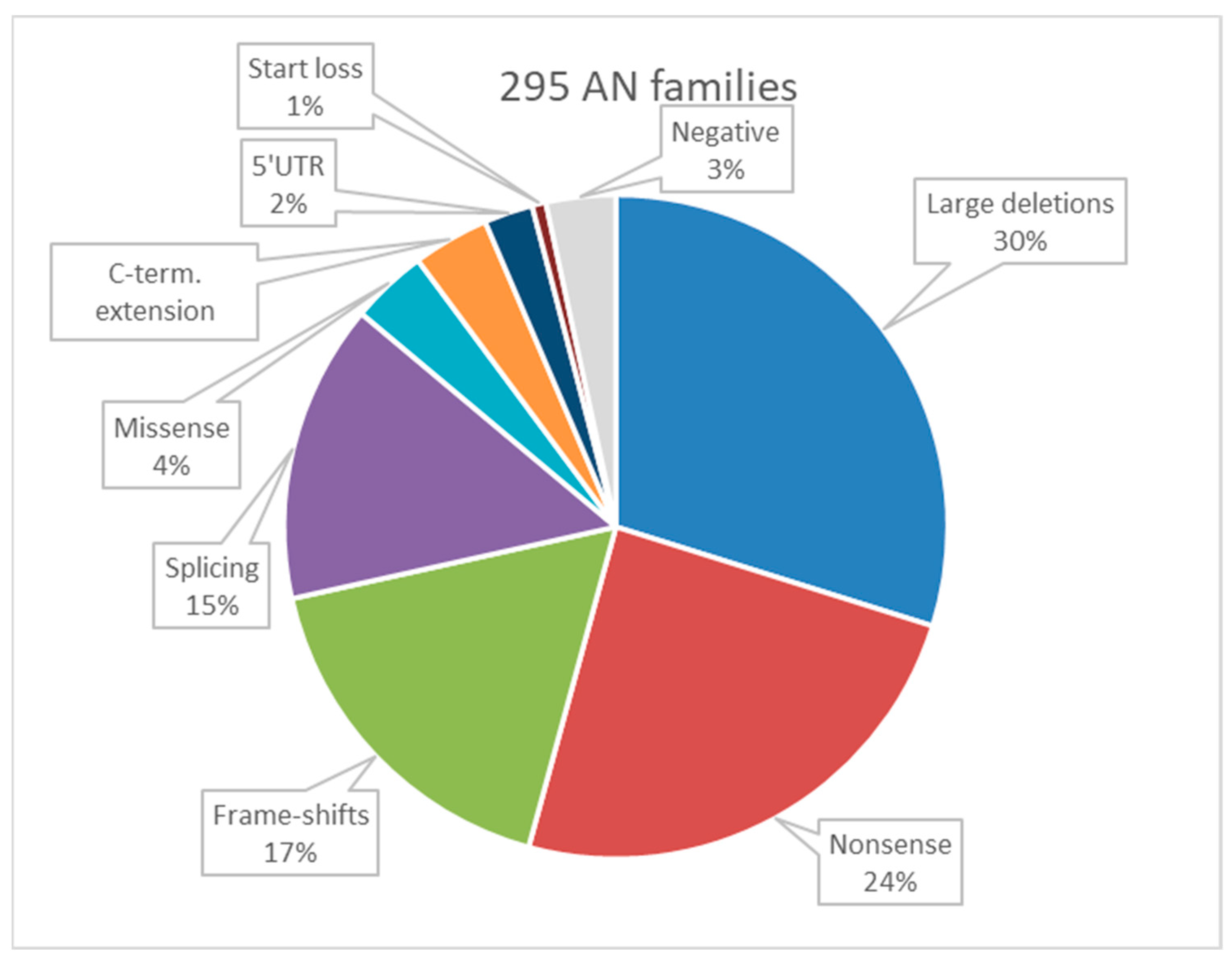
3.3. Summarized Data on PAX6 Variants Defined in a Broadened Sample (All in All 295 Aniridia Families with 379 Patients)
3.4. Expected Number of Patients with Congenital Aniridia across the Russian Federation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hingorani, M.; Williamson, K.A.; Moore, A.T.; van Heyningen, V. Detailed ophthalmologic evaluation of 43 individuals with pax6 mutations. Investig. Ophthalmol. Vis. Sci. 2009, 50, 2581–2590. [Google Scholar] [CrossRef] [PubMed]
- Tzoulaki, I.; White, I.M.; Hanson, I.M. Pax6 mutations: Genotype-phenotype correlations. BMC Genet. 2005, 6, 27. [Google Scholar] [CrossRef] [PubMed]
- Kleinjan, D.A.; Seawright, A.; Mella, S.; Carr, C.B.; Tyas, D.A.; Simpson, T.I.; Mason, J.O.; Price, D.J.; van Heyningen, V. Long-range downstream enhancers are essential for pax6 expression. Dev. Biol. 2006, 299, 563–581. [Google Scholar] [CrossRef]
- Damian, A.; Nunez-Moreno, G.; Jubin, C.; Tamayo, A.; de Alba, M.R.; Villaverde, C.; Fund, C.; Delepine, M.; Leduc, A.; Deleuze, J.F.; et al. Long-read genome sequencing identifies cryptic structural variants in congenital aniridia cases. Hum. Genom. 2023, 17, 45. [Google Scholar] [CrossRef] [PubMed]
- Fischbach, B.V.; Trout, K.L.; Lewis, J.; Luis, C.A.; Sika, M. Wagr syndrome: A clinical review of 54 cases. Pediatrics 2005, 116, 984–988. [Google Scholar] [CrossRef] [PubMed]
- Green, J.S.; Bear, J.C.; Johnson, G.J. The burden of genetically determined eye disease. Br. J. Ophthalmol. 1986, 70, 696–699. [Google Scholar] [CrossRef]
- Das, R.; Savina, E.A.; Tatarinova, T.V.; Orlov, Y.L. Editorial: Population and ancestry specific variation in disease susceptibility. Front. Genet. 2023, 14, 1267719. [Google Scholar] [CrossRef]
- Zinchenko, R.A.; Ginter, E.K.; Marakhonov, A.V.; Petrova, N.V.; Kadyshev, V.V.; Vasilyeva, T.P.; Alexandrova, O.U.; Polyakov, A.V.; Kutsev, S.I. Epidemiology of rare hereditary diseases in the European part of Russia: Point and cumulative prevalence. Front. Genet. 2021, 12, 678957. [Google Scholar] [CrossRef]
- Hamamy, H.; Alwan, A. Hereditary disorders in the eastern mediterranean region. Bull. World Health Org. 1994, 72, 145–154. [Google Scholar]
- Vasilyeva, T.A.; Voskresenskaya, A.A.; Käsmann-Kellner, B.; Khlebnikova, O.V.; Pozdeyeva, N.A.; Bayazutdinova, G.M.; Kutsev, S.I.; Ginter, E.K.; Semina, E.V.; Marakhonov, A.V.; et al. Molecular analysis of patients with aniridia in russian federation broadens the spectrum of pax6 mutations. Clin. Genet. 2017, 92, 639–644. [Google Scholar] [CrossRef]
- Cavalli-Sforza, L.L.; Bodmer, W.F. The Genetics of Human Populations; Dover Publications: Mineola, NY, USA, 1999; 965p. [Google Scholar]
- Abramson, J.H. Winpepi updated: Computer programs for epidemiologists, and their teaching potential. Epidemiol. Perspect. Innov. 2011, 8, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela, A.; Cline, R.A. Ocular and nonocular findings in patients with aniridia. Can. J. Ophthalmol. 2004, 39, 632–638. [Google Scholar] [CrossRef] [PubMed]
- Fries, F.N.; Naray, A.; Munteanu, C.; Stachon, T.; Lagali, N.; Seitz, B.; Szentmary, N.; Kasmann-Kellner, B. A cross-sectional analysis of 556 eyes entering the homburg aniridia centre. Klin. Monbl. Augenheilkd. 2023. [Google Scholar] [CrossRef] [PubMed]
- Heyman, I.; Frampton, I.; van Heyningen, V.; Hanson, I.; Teague, P.; Taylor, A.; Simonoff, E. Psychiatric disorder and cognitive function in a family with an inherited novel mutation of the developmental control gene pax6. Psychiatr. Genet. 1999, 9, 85–90. [Google Scholar] [CrossRef]
- Bamiou, D.E.; Musiek, F.E.; Sisodiya, S.M.; Free, S.L.; Mitchell, T.N.; Davies, R.A. Defective auditory interhemispheric transfer in a patient with a pax6 mutation. Neurology 2004, 62, 489–490. [Google Scholar] [CrossRef]
- Sisodiya, S.M.; Free, S.L.; Williamson, K.A.; Mitchell, T.N.; Willis, C.; Stevens, J.M.; Kendall, B.E.; Shorvon, S.D.; Hanson, I.M.; Moore, A.T.; et al. Pax6 haploinsufficiency causes cerebral malformation and olfactory dysfunction in humans. Nat. Genet. 2001, 28, 214–216. [Google Scholar] [CrossRef]
- Cox, M.A.; Davis, M.; Voin, V.; Shoja, M.; Oskouian, R.J.; Loukas, M.; Tubbs, R.S. Pineal gland agenesis: Review and case illustration. Cureus 2017, 9, e1314. [Google Scholar] [CrossRef]
- Yogarajah, M.; Matarin, M.; Vollmar, C.; Thompson, P.J.; Duncan, J.S.; Symms, M.; Moore, A.T.; Liu, J.; Thom, M.; van Heyningen, V.; et al. Pax6, brain structure and function in human adults: Advanced mri in aniridia. Ann. Clin. Transl. Neurol. 2016, 3, 314–330. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the american college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Vasilyeva, T.A.; Marakhonov, A.V.; Minzhenkova, M.E.; Markova, Z.G.; Petrova, N.V.; Sukhanova, N.V.; Koshkin, P.A.; Pyankov, D.V.; Kanivets, I.V.; Korostelev, S.A.; et al. A sporadic case of congenital aniridia caused by pericentric inversion inv(11)(p13q14) associated with a 977 kb deletion in the 11p13 region. BMC Med. Genom. 2020, 13, 130. [Google Scholar] [CrossRef]
- Orphadata. Prevalence of Rare Diseases: Bibliographic Data—January 2019; Orphadata: Paris, France, 2019. [Google Scholar]
- Eden, U.; Iggman, D.; Riise, R.; Tornqvist, K. Epidemiology of aniridia in sweden and norway. Acta Ophthalmol. 2008, 86, 727–729. [Google Scholar] [CrossRef] [PubMed]
- Kadyshev, V.V.; Ginter, E.K.; Kutsev, S.I.; Oganezova, Z.G.; Zinchenko, R.A. Epidemiology of hereditary eye disease in the populations of russian federation. Rus. J. Clin. Ophthalmol. 2022, 22, 69–79. (In Russian) [Google Scholar] [CrossRef]
- Kim, J.; Lauderdale, J.D. Analysis of pax6 expression using a bac transgene reveals the presence of a paired-less isoform of pax6 in the eye and olfactory bulb. Dev. Biol. 2006, 292, 486–505. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Aniridia Features | Defined in N Patients | Out of the Studied N Patients with Data on the Feature | Portion of the Patients with the Feature Defined in the Study | Portion of the Patients with the Feature Defined Earlier |
|---|---|---|---|---|
| Complete absence of iris | 256 | 318 | 80.5% | 78% |
| Partial absence of iris/iris hypoplasia/coloboma | 62 | 318 | 19.5% | 22% |
| Cataract | 247 | 311 | 79.4% | 80% |
| Foveal hypoplasia | 244 | 264 | 92.4% | 85% |
| Keratopathy | 182 | 307 | 59.3% | 58% |
| Nystagmus | 235 | 280 | 83.9% | 78% |
| Glaucoma | 78 | 318 | 24.6% | 26% |
| Patient | Karyotype | Visible Event | Additional Events | Deletion in 11p13 (hg18) |
|---|---|---|---|---|
| t-384.02 | 46,XY | t(10,11)(p15;p13) | inv(11)(p12q12) | g.(31285887_35117390)del |
| t-509.04 | 46,XX | t(2;11)(q34;p15) | nd 1 | nd 1 |
| t-92.03 | 46,XY | inv(11)(p13q14) | inv(11)(p14p13) + 11q23.3_11q25.1del | nd 1 |
| A-36 | 46,XY | inv(11)(p14p13) | nd 1 | g.(31628232_32296427)del |
| District | Age-Adjusted Population Size (under 65 y.o.) | Observed Number of AN Cases (N) | Expected Number of AN Cases (Based on Established Prevalence, N*) | Ratio of Out-of-Sight Cases (1 − N/N*) |
|---|---|---|---|---|
| Northwestern | 11 648 915 | 24 | 118 | 0.80 |
| Central | 33 793 164 | 85 | 342 | 0.75 |
| Volga | 24 094 401 | 110 | 244 | 0.55 |
| Southern | 14 043 291 | 33 | 142 | 0.77 |
| North Caucasian | 8 571 229 | 27 | 87 | 0.69 |
| Ural | 10 294 912 | 40 | 104 | 0.62 |
| Siberian | 13 982 372 | 23 | 141 | 0.84 |
| Far Eastern | 6 638 194 | 9 | 67 | 0.87 |
| Total | 123 066 483 | 351 | 1244 | 0.72 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vasilyeva, T.A.; Marakhonov, A.V.; Voskresenskaya, A.A.; Kadyshev, V.V.; Sukhanova, N.V.; Minzhenkova, M.E.; Shilova, N.V.; Latyshova, A.A.; Ginter, E.K.; Kutsev, S.I.; et al. Epidemiology of PAX6 Gene Pathogenic Variants and Expected Prevalence of PAX6-Associated Congenital Aniridia across the Russian Federation: A Nationwide Study. Genes 2023, 14, 2041. https://doi.org/10.3390/genes14112041
Vasilyeva TA, Marakhonov AV, Voskresenskaya AA, Kadyshev VV, Sukhanova NV, Minzhenkova ME, Shilova NV, Latyshova AA, Ginter EK, Kutsev SI, et al. Epidemiology of PAX6 Gene Pathogenic Variants and Expected Prevalence of PAX6-Associated Congenital Aniridia across the Russian Federation: A Nationwide Study. Genes. 2023; 14(11):2041. https://doi.org/10.3390/genes14112041
Chicago/Turabian StyleVasilyeva, Tatyana A., Andrey V. Marakhonov, Anna A. Voskresenskaya, Vitaly V. Kadyshev, Natella V. Sukhanova, Marina E. Minzhenkova, Nadezhda V. Shilova, Alla A. Latyshova, Evgeny K. Ginter, Sergey I. Kutsev, and et al. 2023. "Epidemiology of PAX6 Gene Pathogenic Variants and Expected Prevalence of PAX6-Associated Congenital Aniridia across the Russian Federation: A Nationwide Study" Genes 14, no. 11: 2041. https://doi.org/10.3390/genes14112041