
Contribution of LRP1 in Human Congenital Heart Disease Correlates with Its Roles in the Outflow Tract and Atrioventricular Cushion Development

 , and
, and 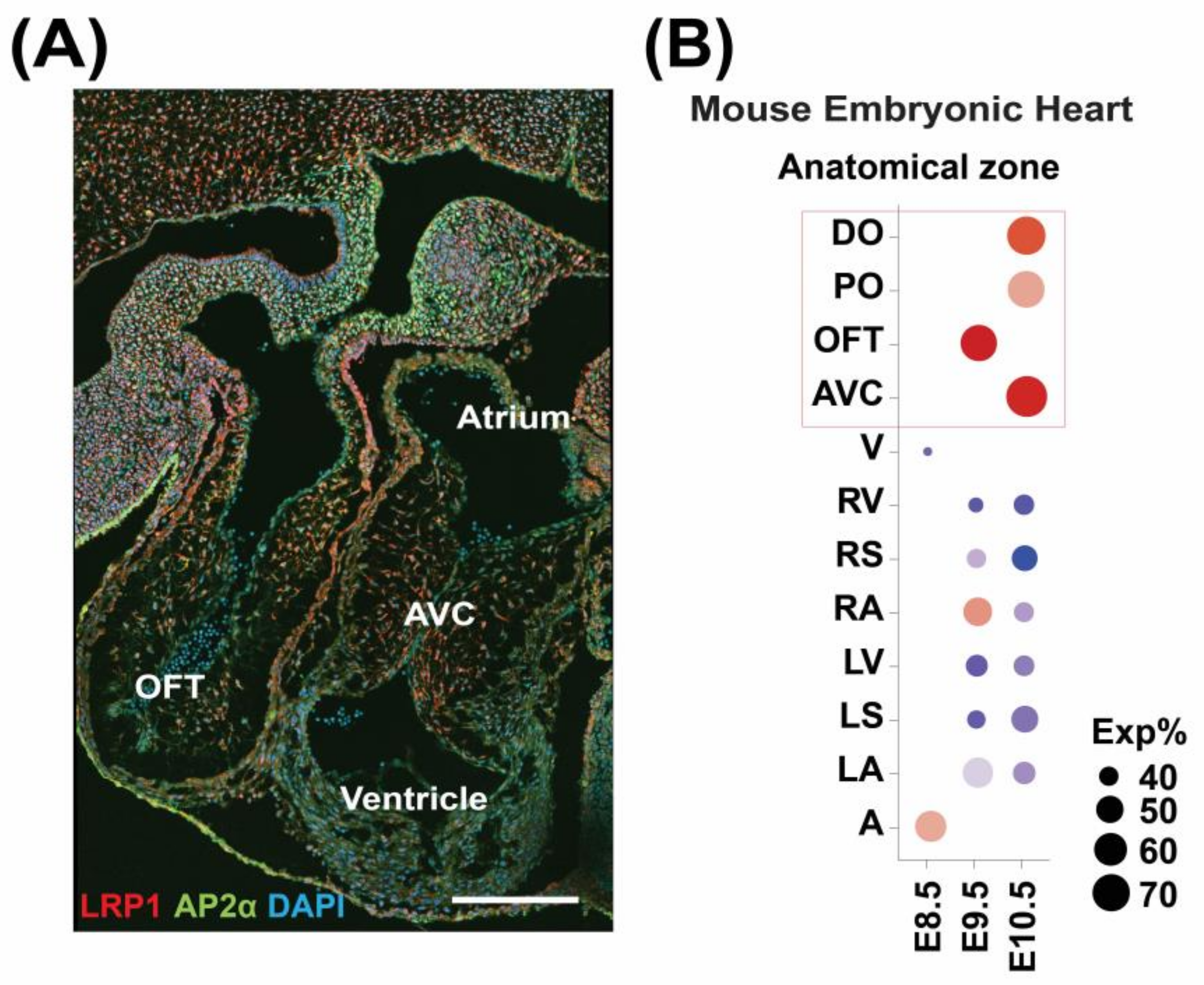{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Immunostaining
2.2. Analysis of Publicly Available Single-Cell RNA Sequencing Data
2.3. Human Study Participants
2.4. Recovery of Rare Predicated Pathogenic LRP1 Variants
2.5. Gene-Based Burden Testing
3. Results
3.1. LRP1 Is Expressed in the Developing Cardiac OFT and AVC
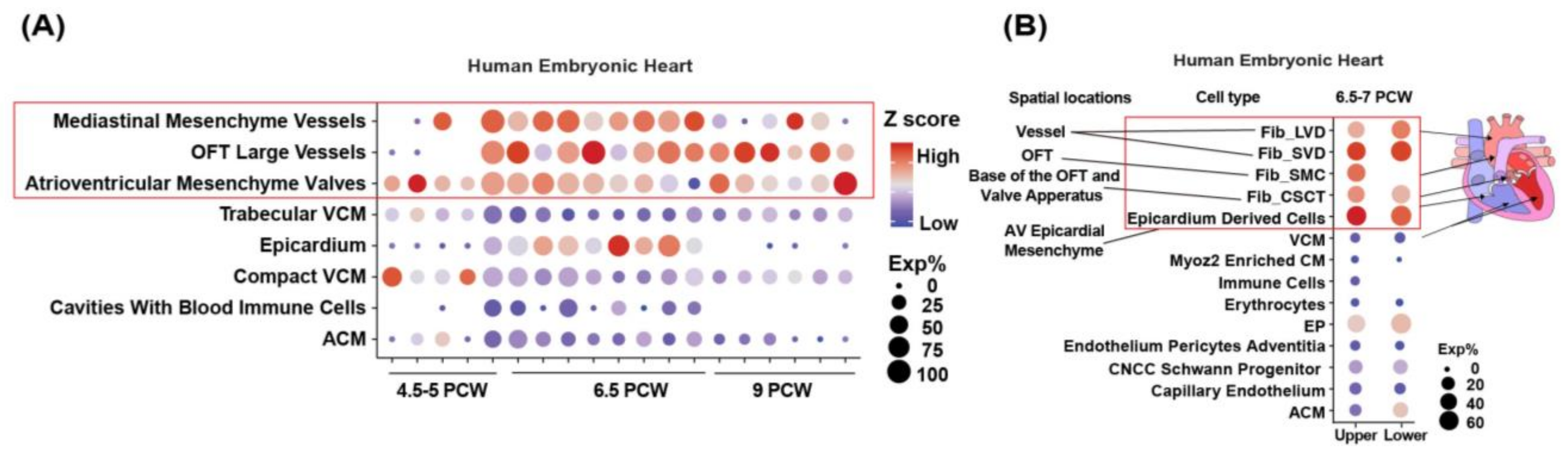
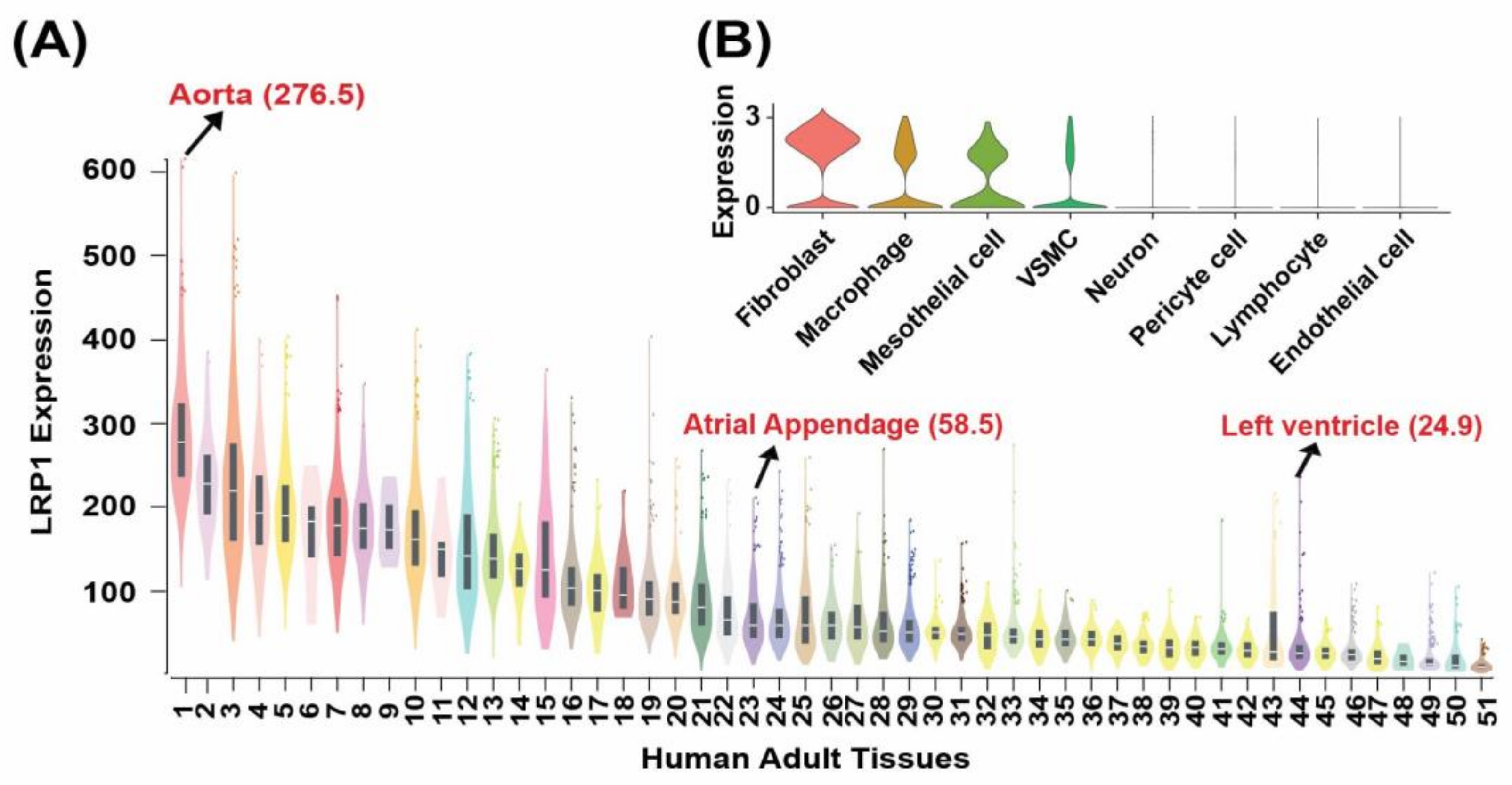
3.2. LRP1 Is Highly Expressed in the Human Aorta
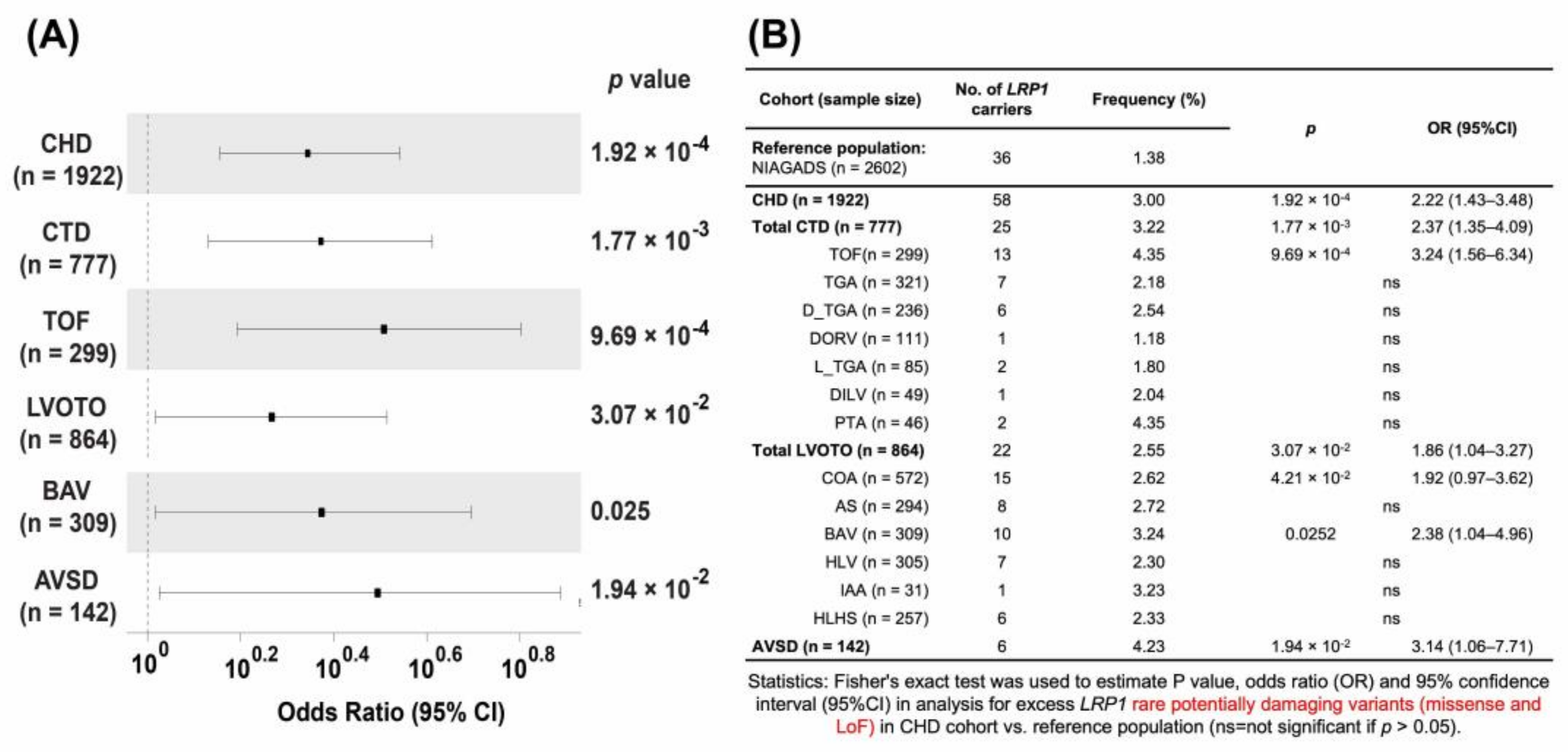
3.3. Nonsynonymous, Rare, and Putative Damaging Variants in LRP1 Are Significantly Associated with CHD
3.4. Rare Potentially Pathogenetic Variants (PPV) Are Enriched in CHD
3.5. Distribution of Rare Damaging Variants in LRP1
3.6. Lrp1 Variants in Patients with AVSD Are Significantly Associated with Their Cases
4. Discussion
5. Conclusions
6. Limitations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lin, J.I.; Feinstein, T.N.; Jha, A.; McCleary, J.T.; Xu, J.; Arrigo, A.B.; Rong, G.; Maclay, L.M.; Ridge, T.; Xu, X.; et al. Mutation of LRP1 in cardiac neural crest cells causes congenital heart defects by perturbing outflow lengthening. Commun. Biol. 2020, 3, 312. [Google Scholar] [CrossRef] [PubMed]
- Hoang, T.T.; Goldmuntz, E.; Roberts, A.E.; Chung, W.K.; Kline, J.K.; Deanfield, J.E.; Giardini, A.; Aleman, A.; Gelb, B.D.; Mac Neal, M.; et al. The Congenital Heart Disease Genetic Network Study: Cohort description. PLoS ONE 2018, 13, e0191319. [Google Scholar] [CrossRef] [PubMed]
- Beecham, G.W.; Bis, J.C.; Martin, E.R.; Choi, S.H.; DeStefano, A.L.; van Duijn, C.M.; Fornage, M.; Gabriel, S.B.; Koboldt, D.C.; Larson, D.E.; et al. The Alzheimer’s Disease Sequencing Project: Study design and sample selection. Neurol. Genet. 2017, 3, e194. [Google Scholar] [CrossRef]
- Li, G.; Xu, A.; Sim, S.; Priest, J.R.; Tian, X.; Khan, T.; Quertermous, T.; Zhou, B.; Tsao, P.S.; Quake, S.R.; et al. Transcriptomic Profiling Maps Anatomically Patterned Subpopulations among Single Embryonic Cardiac Cells. Dev. Cell 2016, 39, 491–507. [Google Scholar] [CrossRef] [PubMed]
- Asp, M.; Giacomello, S.; Larsson, L.; Wu, C.; Furth, D.; Qian, X.; Wardell, E.; Custodio, J.; Reimegard, J.; Salmen, F.; et al. A Spatiotemporal Organ-Wide Gene Expression and Cell Atlas of the Developing Human Heart. Cell 2019, 179, 1647–1660.e19. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Williams, K.; Young, C.; Lin, J.H.; Teekakirikul, P.; Lo, C.W. Rare and Common Variants Uncover the Role of the Atria in Coarctation of the Aorta. Genes 2022, 13, 636. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows—Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef]
- Pruitt, K.D.; Tatusova, T.; Maglott, D.R. NCBI reference sequences (RefSeq): A curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2007, 35, D61–D65. [Google Scholar] [CrossRef] [PubMed]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Hoover, J.; et al. ClinVar: Public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016, 44, D862–D868. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 76, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Jun, G.; Flickinger, M.; Hetrick, K.N.; Romm, J.M.; Doheny, K.F.; Abecasis, G.R.; Boehnke, M.; Kang, H.M. Detecting and estimating contamination of human DNA samples in sequencing and array-based genotype data. Am. J. Hum. Genet. 2012, 91, 839–848. [Google Scholar] [CrossRef] [PubMed]
- Manichaikul, A.; Mychaleckyj, J.C.; Rich, S.S.; Daly, K.; Sale, M.; Chen, W.M. Robust relationship inference in genome-wide association studies. Bioinformatics 2010, 26, 2867–2873. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Walsh, R.; Thomson, K.L.; Ware, J.S.; Funke, B.H.; Woodley, J.; McGuire, K.J.; Mazzarotto, F.; Blair, E.; Seller, A.; Taylor, J.C.; et al. Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genet. Med. 2017, 19, 192–203. [Google Scholar] [CrossRef]
- Walsh, R.; Buchan, R.; Wilk, A.; John, S.; Felkin, L.E.; Thomson, K.L.; Chiaw, T.H.; Loong, C.C.W.; Pua, C.J.; Raphael, C.; et al. Defining the genetic architecture of hypertrophic cardiomyopathy: Re-evaluating the role of non-sarcomeric genes. Eur. Heart J. 2017, 38, 3461–3468. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.H.; Plummer, L.; Chan, Y.-M.; Hirschhorn, J.N.; Lippincott, M.F. Burden Testing of Rare Variants Identified through Exome Sequencing via Publicly Available Control Data. Am. J. Hum. Genet 2018, 103, 522–534. [Google Scholar] [CrossRef]
- GTEx Consortium. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef] [PubMed]
- GTEx Consortium. The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science 2015, 348, 648–660. [Google Scholar] [CrossRef]
- Pirruccello, J.P.; Chaffin, M.D.; Chou, E.L.; Fleming, S.J.; Lin, H.; Nekoui, M.; Khurshid, S.; Friedman, S.F.; Bick, A.G.; Arduini, A.; et al. Deep learning enables genetic analysis of the human thoracic aorta. Nat. Genet. 2022, 54, 40–51. [Google Scholar] [CrossRef]
- Gonias, S.L. Mechanisms by Which LRP1 (Low-Density Lipoprotein Receptor–Related Protein-1) Maintains Arterial Integrity. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 2548–2549. [Google Scholar] [CrossRef] [PubMed]
- Bis, J.C.; Jian, X.; Kunkle, B.W.; Chen, Y.; Hamilton-Nelson, K.L.; Bush, W.S.; Salerno, W.J.; Lancour, D.; Ma, Y.; Renton, A.E.; et al. Whole exome sequencing study identifies novel rare and common Alzheimer’s-Associated variants involved in immune response and transcriptional regulation. Mol. Psychiatry 2020, 25, 1859–1875. [Google Scholar] [CrossRef] [PubMed]
- Baardman, M.E.; Zwier, M.V.; Wisse, L.J.; Gittenberger-de Groot, A.C.; Kerstjens-Frederikse, W.S.; Hofstra, R.M.; Jurdzinski, A.; Hierck, B.P.; Jongbloed, M.R.; Berger, R.M.; et al. Common arterial trunk and ventricular non-compaction in Lrp2 knockout mice indicate a crucial role of LRP2 in cardiac development. Dis. Model. Mech. 2016, 9, 413–425. [Google Scholar] [CrossRef]
- Bansal, A.; Gierasch, L.M. The NPXY internalization signal of the LDL receptor adopts a reverse-turn conformation. Cell 1991, 67, 1195–1201. [Google Scholar] [CrossRef]
- Strickland, D.K.; Au, D.T.; Cunfer, P.; Muratoglu, S.C. Low-density lipoprotein receptor-related protein-1: Role in the regulation of vascular integrity. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 487–498. [Google Scholar] [CrossRef]
- Potere, N.; Del Buono, M.G.; Niccoli, G.; Crea, F.; Toldo, S.; Abbate, A. Developing LRP1 Agonists into a Therapeutic Strategy in Acute Myocardial Infarction. Int. J. Mol. Sci. 2019, 20, 544. [Google Scholar] [CrossRef] [PubMed]
- Herz, J.; Clouthier, D.E.; Hammer, R.E. LDL receptor-related protein internalizes and degrades uPA-PAI-1 complexes and is essential for embryo implantation. Cell 1992, 71, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Klena, N.T.; Gabriel, G.C.; Liu, X.; Kim, A.J.; Lemke, K.; Chen, Y.; Chatterjee, B.; Devine, W.; Damerla, R.R.; et al. Global genetic analysis in mice unveils central role for cilia in congenital heart disease. Nature 2015, 521, 520–524. [Google Scholar] [CrossRef] [PubMed]
- Pi, X.; Schmitt, C.E.; Xie, L.; Portbury, A.L.; Wu, Y.; Lockyer, P.; Dyer, L.A.; Moser, M.; Bu, G.; Flynn, E.J., 3rd; et al. LRP1-dependent endocytic mechanism governs the signaling output of the bmp system in endothelial cells and in angiogenesis. Circ. Res. 2012, 111, 564–574. [Google Scholar] [CrossRef] [PubMed]
- Bian, W.; Tang, M.; Jiang, H.; Xu, W.; Hao, W.; Sui, Y.; Hou, Y.; Nie, L.; Zhang, H.; Wang, C.; et al. Low-density-lipoprotein-receptor-related protein 1 mediates Notch pathway activation. Dev. Cell 2021, 56, 2902–2919.e8. [Google Scholar] [CrossRef]
- Beloglazova, I.; Stepanova, V.; Zubkova, E.; Dergilev, K.; Koptelova, N.; Tyurin-Kuzmin, P.A.; Dyikanov, D.; Plekhanova, O.; Cines, D.B.; Mazar, A.P.; et al. Mesenchymal stromal cells enhance self-assembly of a HUVEC tubular network through uPA-uPAR/VEGFR2/integrin/NOTCH crosstalk. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2022, 1869, 119157. [Google Scholar] [CrossRef]
- Meng, H.; Zhang, X.; Lee, S.J.; Strickland, D.K.; Lawrence, D.A.; Wang, M.M. Low density lipoprotein receptor-related protein-1 (LRP1) regulates thrombospondin-2 (TSP2) enhancement of Notch3 signaling. J. Biol. Chem. 2010, 285, 23047–23055. [Google Scholar] [CrossRef]
- Terrand, J.; Bruban, V.; Zhou, L.; Gong, W.; El Asmar, Z.; May, P.; Zurhove, K.; Haffner, P.; Philippe, C.; Woldt, E.; et al. LRP1 controls intracellular cholesterol storage and fatty acid synthesis through modulation of Wnt signaling. J. Biol. Chem. 2009, 284, 381–388. [Google Scholar] [CrossRef]
- Au, D.T.; Migliorini, M.; Strickland, D.K.; Muratoglu, S.C. Macrophage LRP1 Promotes Diet-Induced Hepatic Inflammation and Metabolic Dysfunction by Modulating Wnt Signaling. Mediat. Inflamm. 2018, 2018, 7902841. [Google Scholar] [CrossRef]
- Hu, K.; Wu, C.; Mars, W.M.; Liu, Y. Tissue-type plasminogen activator promotes murine myofibroblast activation through LDL receptor-related protein 1-mediated integrin signaling. J. Clin. Investig. 2007, 117, 3821–3832. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arrigo, A.B.; Zhu, W.; Williams, K.A.; Guzman-Moreno, C.; Lo, C.; Lin, J.-H.I. Contribution of LRP1 in Human Congenital Heart Disease Correlates with Its Roles in the Outflow Tract and Atrioventricular Cushion Development. Genes 2023, 14, 947. https://doi.org/10.3390/genes14040947
Arrigo AB, Zhu W, Williams KA, Guzman-Moreno C, Lo C, Lin J-HI. Contribution of LRP1 in Human Congenital Heart Disease Correlates with Its Roles in the Outflow Tract and Atrioventricular Cushion Development. Genes. 2023; 14(4):947. https://doi.org/10.3390/genes14040947
Chicago/Turabian StyleArrigo, Angelo B., Wenjuan Zhu, Kylia A. Williams, Carla Guzman-Moreno, Cecilia Lo, and Jiuann-Huey I. Lin. 2023. "Contribution of LRP1 in Human Congenital Heart Disease Correlates with Its Roles in the Outflow Tract and Atrioventricular Cushion Development" Genes 14, no. 4: 947. https://doi.org/10.3390/genes14040947