
Analysis of Viral Promoters for Transgene Expression and of the Effect of 5′-UTRs on Alternative Translational Start Sites in Chlamydomonas

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Strains and Culture Conditions
2.2. Nuclear Transformation of Chlamydomonas
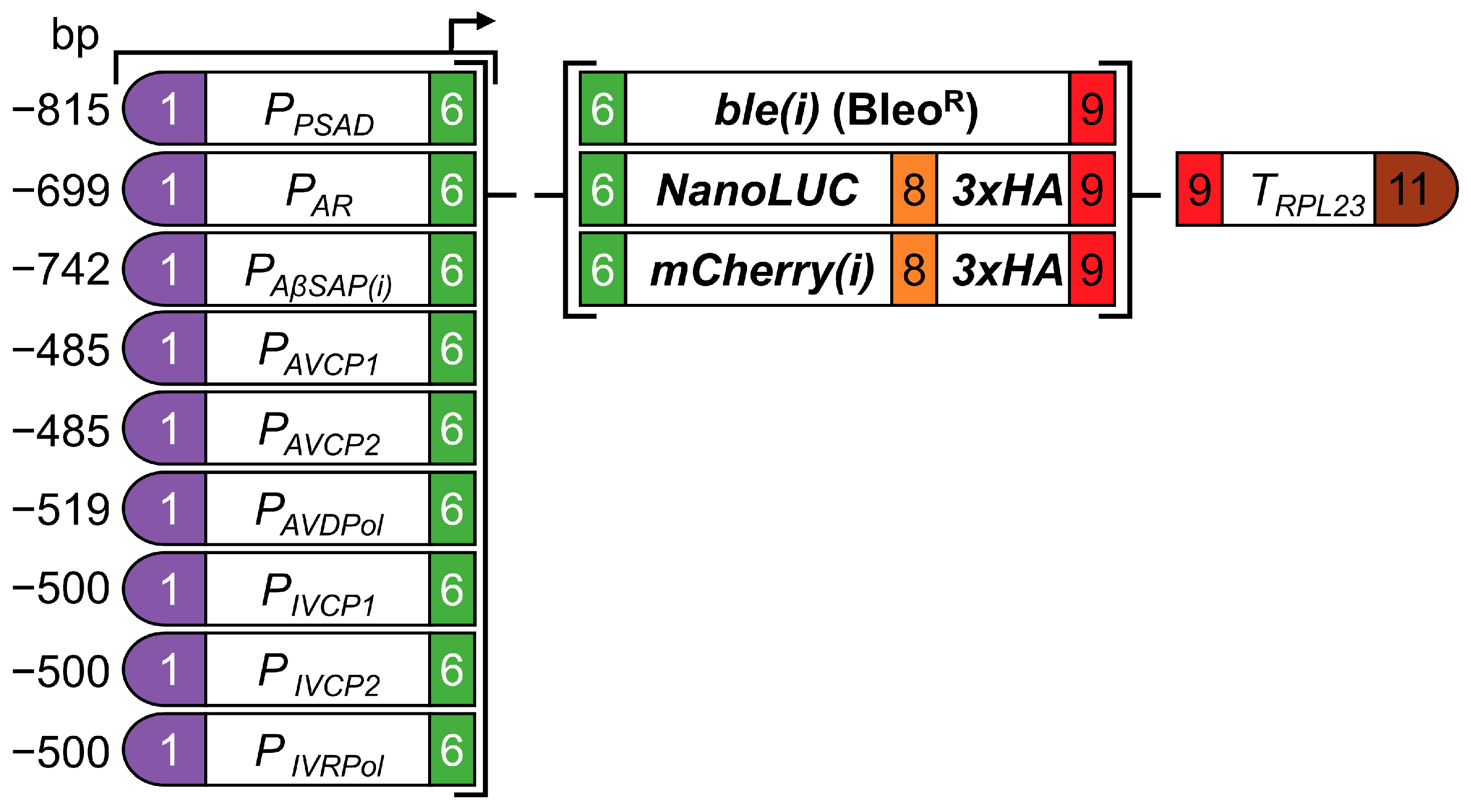
2.3. Cloning
2.4. Determination of NanoLUC Activity
2.5. Determination of mCherry Activity
2.6. Protein Analyses
3. Results
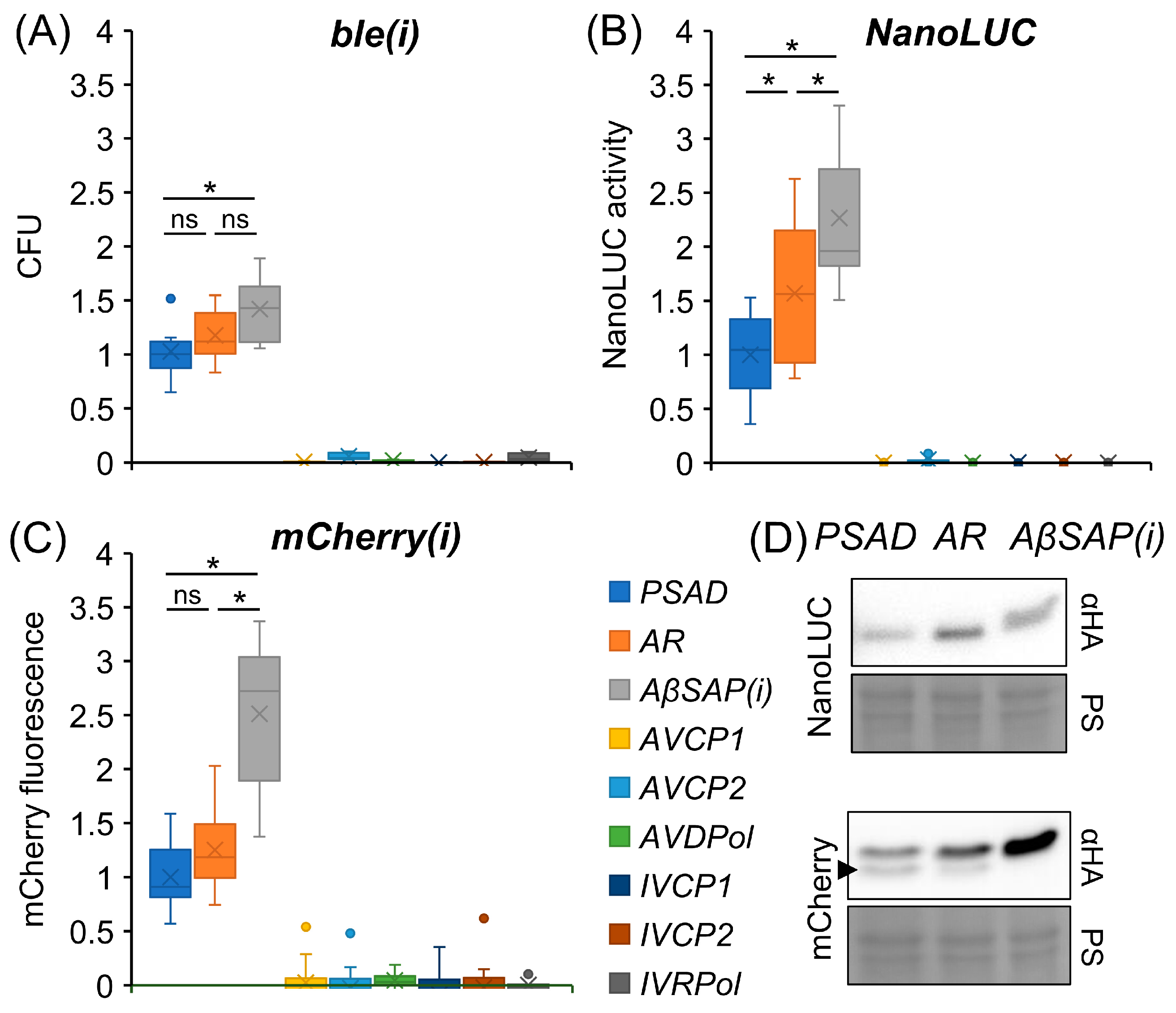
3.1. Viral Promoters Are Not Functional in Chlamydomonas
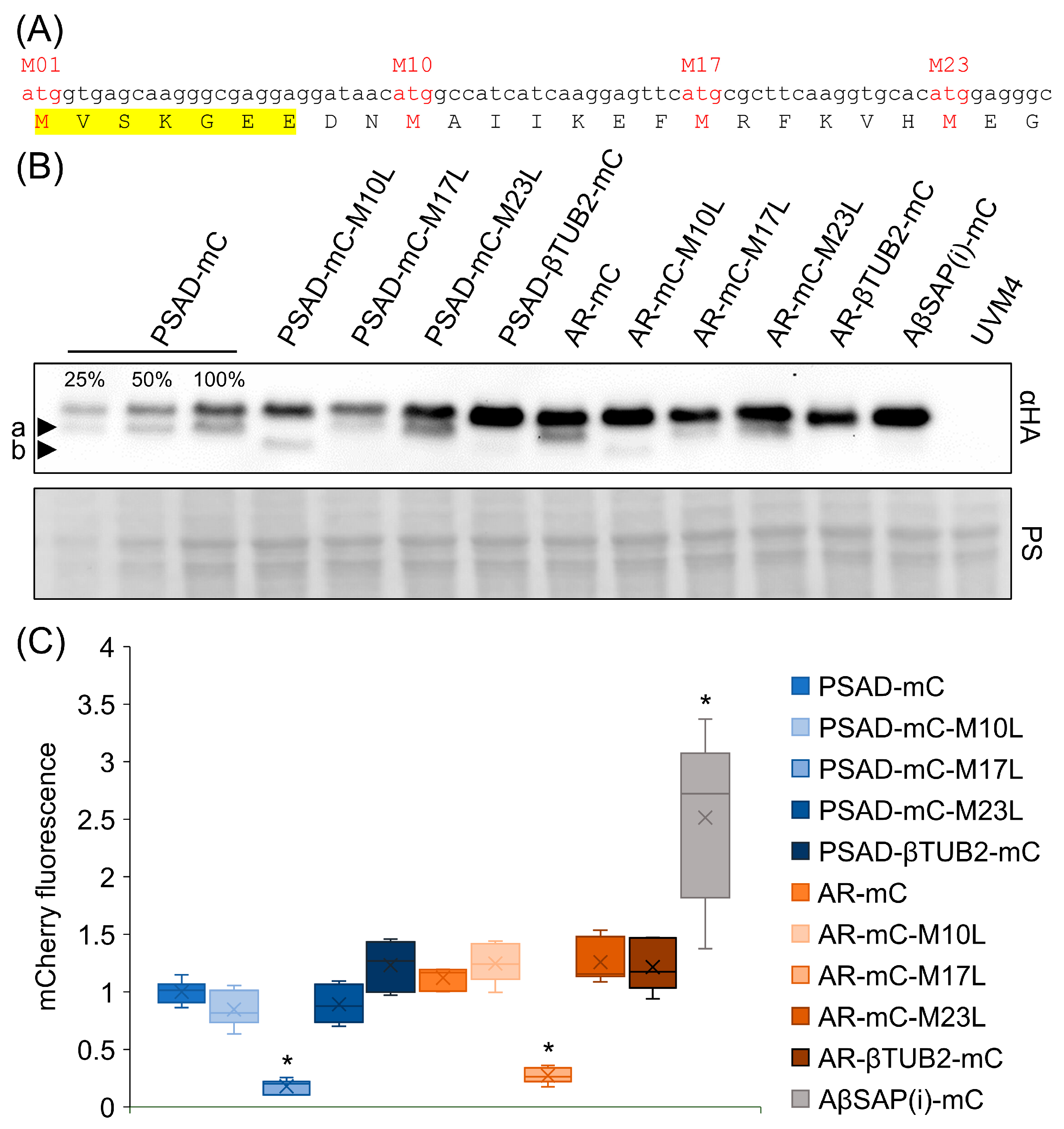
3.2. The βTUB2 5′-UTR Prevents the Production of a Shorter mCherry Isoform
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Berndt, A.J.; Smalley, T.N.; Ren, B.; Simkovsky, R.; Badary, A.; Sproles, A.E.; Fields, F.J.; Torres-Tiji, Y.; Heredia, V.; Mayfield, S.P. Recombinant production of a functional SARS-CoV-2 spike receptor binding domain in the green algae Chlamydomonas reinhardtii. PLoS ONE 2021, 16, e0257089. [Google Scholar] [CrossRef] [PubMed]
- Einhaus, A.; Baier, T.; Rosenstengel, M.; Freudenberg, R.A.; Kruse, O. Rational promoter engineering enables robust terpene production in microalgae. ACS Synth. Biol. 2021, 10, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Kiefer, A.M.; Niemeyer, J.; Probst, A.; Erkel, G.; Schroda, M. Production and secretion of functional SARS-CoV-2 spike protein in Chlamydomonas reinhardtii. Front. Plant Sci. 2022, 13, 988870. [Google Scholar] [CrossRef] [PubMed]
- Overmans, S.; Lauersen, K.J. Biocompatible fluorocarbon liquid underlays for in situ extraction of isoprenoids from microbial cultures. RSC Adv. 2022, 12, 16632–16639. [Google Scholar] [CrossRef]
- Wichmann, J.; Eggert, A.; Elbourne, L.D.H.; Paulsen, I.T.; Lauersen, K.J.; Kruse, O. Farnesyl pyrophosphate compartmentalization in the green microalga Chlamydomonas reinhardtii during heterologous (E)-alpha-bisabolene production. Microb. Cell Fact. 2022, 21, 190. [Google Scholar] [CrossRef]
- Schroda, M.; Remacle, C. Molecular advancements establishing Chlamydomonas as a host for biotechnological exploitation. Front. Plant Sci. 2022, 13, 911483. [Google Scholar] [CrossRef]
- Scaife, M.A.; Nguyen, G.T.; Rico, J.; Lambert, D.; Helliwell, K.E.; Smith, A.G. Establishing Chlamydomonas reinhardtii as an industrial biotechnology host. Plant J. 2015, 82, 532–546. [Google Scholar] [CrossRef]
- Baier, T.; Wichmann, J.; Kruse, O.; Lauersen, K.J. Intron-containing algal transgenes mediate efficient recombinant gene expression in the green microalga Chlamydomonas reinhardtii. Nucleic Acids Res. 2018, 46, 6909–6919. [Google Scholar] [CrossRef]
- Barahimipour, R.; Strenkert, D.; Neupert, J.; Schroda, M.; Merchant, S.S.; Bock, R. Dissecting the contributions of GC content and codon usage to gene expression in the model alga Chlamydomonas reinhardtii. Plant J. 2015, 84, 704–717. [Google Scholar] [CrossRef]
- Neupert, J.; Gallaher, S.D.; Lu, Y.; Strenkert, D.; Segal, N.; Barahimipour, R.; Fitz-Gibbon, S.T.; Schroda, M.; Merchant, S.S.; Bock, R. An epigenetic gene silencing pathway selectively acting on transgenic DNA in the green alga Chlamydomonas. Nat. Commun. 2020, 11, 6269. [Google Scholar] [CrossRef]
- Strenkert, D.; Schmollinger, S.; Schroda, M. Heat shock factor 1 counteracts epigenetic silencing of nuclear transgenes in Chlamydomonas reinhardtii. Nucleic Acids Res. 2013, 41, 5273–5289. [Google Scholar] [CrossRef] [PubMed]
- Schroda, M. Good news for nuclear transgene expression in Chlamydomonas. Cells 2019, 8, 1534. [Google Scholar] [CrossRef] [PubMed]
- Fischer, N.; Rochaix, J.D. The flanking regions of PsaD drive efficient gene expression in the nucleus of the green alga Chlamydomonas reinhardtii. Mol. Genet. Genom. MGG 2001, 265, 888–894. [Google Scholar] [CrossRef] [PubMed]
- Schroda, M.; Blocker, D.; Beck, C.F. The HSP70A promoter as a tool for the improved expression of transgenes in Chlamydomonas. Plant J. 2000, 21, 121–131. [Google Scholar] [CrossRef]
- Stevens, D.R.; Rochaix, J.D.; Purton, S. The bacterial phleomycin resistance gene ble as a dominant selectable marker in Chlamydomonas. Mol. Gen. Genet. 1996, 251, 23–30. [Google Scholar]
- Blankenship, J.E.; Kindle, K.L. Expression of chimeric genes by the light-regulated cabII-1 promoter in Chlamydomonas reinhardtii: A cabII-1/nit1 gene functions as a dominant selectable marker in a nit1-nit2-strain. Mol. Cell. Biol. 1992, 12, 5268–5279. [Google Scholar] [CrossRef]
- Ruecker, O.; Zillner, K.; Groebner-Ferreira, R.; Heitzer, M. Gaussia-luciferase as a sensitive reporter gene for monitoring promoter activity in the nucleus of the green alga Chlamydomonas reinhardtii. Mol. Genet. Genom. MGG 2008, 280, 153–162. [Google Scholar] [CrossRef]
- Day, A.; Debuchy, R.; Vandillewijn, J.; Purton, S.; Rochaix, J.D. Studies on the Maintenance and Expression of Cloned DNA Fragments in the Nuclear Genome of the Green Alga Chlamydomonas reinhardtii. Physiol. Plant. 1990, 78, 254–260. [Google Scholar] [CrossRef]
- Díaz-Santos, E.; de la Vega, M.; Vila, M.; Vigara, J.; León, R. Efficiency of different heterologous promoters in the unicellular microalga Chlamydomonas reinhardtii. Biotechnol. Prog. 2013, 29, 319–328. [Google Scholar] [CrossRef]
- Dong, B.; Hu, H.H.; Li, Z.F.; Cheng, R.Q.; Meng, D.M.; Wang, J.; Fan, Z.C. A novel bicistronic expression system composed of the intraflagellar transport protein gene ift25 and FMDV 2A sequence directs robust nuclear gene expression in Chlamydomonas reinhardtii. Appl. Microbiol. Biotechnol. 2017, 101, 4227–4245. [Google Scholar] [CrossRef]
- Moniruzzaman, M.; Erazo-Garcia, M.P.; Aylward, F.O. Endogenous giant viruses contribute to intraspecies genomic variability in the model green alga Chlamydomonas reinhardtii. Virus Evol. 2022, 8, veac102. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Yutin, N. Evolution of the Large Nucleocytoplasmic DNA Viruses of Eukaryotes and Convergent Origins of Viral Gigantism. Adv. Virus Res. 2019, 103, 167–202. [Google Scholar] [CrossRef] [PubMed]
- Yutin, N.; Wolf, Y.I.; Koonin, E.V. Origin of giant viruses from smaller DNA viruses not from a fourth domain of cellular life. Virology 2014, 466–467, 38–52. [Google Scholar] [CrossRef] [PubMed]
- Aylward, F.O.; Moniruzzaman, M.; Ha, A.D.; Koonin, E.V. A phylogenomic framework for charting the diversity and evolution of giant viruses. PLoS Biol. 2021, 19, e3001430. [Google Scholar] [CrossRef] [PubMed]
- Moniruzzaman, M.; Weinheimer, A.R.; Martinez-Gutierrez, C.A.; Aylward, F.O. Widespread endogenization of giant viruses shapes genomes of green algae. Nature 2020, 588, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Merchant, S.S.; Prochnik, S.E.; Vallon, O.; Harris, E.H.; Karpowicz, S.J.; Witman, G.B.; Terry, A.; Salamov, A.; Fritz-Laylin, L.K.; Marechal-Drouard, L.; et al. The Chlamydomonas genome reveals the evolution of key animal and plant functions. Science 2007, 318, 245–250. [Google Scholar] [CrossRef]
- Neupert, J.; Karcher, D.; Bock, R. Generation of Chlamydomonas strains that efficiently express nuclear transgenes. Plant J. 2009, 57, 1140–1150. [Google Scholar] [CrossRef]
- Kropat, J.; Hong-Hermesdorf, A.; Casero, D.; Ent, P.; Castruita, M.; Pellegrini, M.; Merchant, S.S.; Malasarn, D. A revised mineral nutrient supplement increases biomass and growth rate in Chlamydomonas reinhardtii. Plant J. 2011, 66, 770–780. [Google Scholar] [CrossRef]
- Kindle, K.L. High-frequency nuclear transformation of Chlamydomonas reinhardtii. Proc. Natl. Acad. Sci. USA 1990, 87, 1228–1232. [Google Scholar] [CrossRef]
- Crozet, P.; Navarro, F.J.; Willmund, F.; Mehrshahi, P.; Bakowski, K.; Lauersen, K.J.; Perez-Perez, M.E.; Auroy, P.; Gorchs Rovira, A.; Sauret-Gueto, S.; et al. Birth of a photosynthetic chassis: A MoClo toolkit enabling Synthetic Biology in the microalga Chlamydomonas reinhardtii. ACS Synth. Biol. 2018, 7, 2074–2086. [Google Scholar] [CrossRef]
- Weber, E.; Engler, C.; Gruetzner, R.; Werner, S.; Marillonnet, S. A modular cloning system for standardized assembly of multigene constructs. PLoS ONE 2011, 6, e16765. [Google Scholar] [CrossRef]
- Niemeyer, J.; Schroda, M. New destination vectors facilitate Modular Cloning for Chlamydomonas. Curr. Genet. 2022, 68, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Porra, R.J.; Thompson, W.A.; Kriedemann, P.E. Determination of accurate extinction coefficients and simultaneous-equations for assaying chlorophyll-a and chlorophyll-b extracted with 4 different solvents—Verification of the concentration of chlorophyll standards by atomic-absorption spectroscopy. Biochim. Biophys. Acta 1989, 975, 384–394. [Google Scholar] [CrossRef]
- Gatignol, A.; Durand, H.; Tiraby, G. Bleomycin resistance conferred by a drug-binding protein. FEBS Lett. 1988, 230, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Schroda, M.; Beck, C.F.; Vallon, O. Sequence elements within an HSP70 promoter counteract transcriptional transgene silencing in Chlamydomonas. Plant J. 2002, 31, 445–455. [Google Scholar] [CrossRef]
- Hall, M.P.; Unch, J.; Binkowski, B.F.; Valley, M.P.; Butler, B.L.; Wood, M.G.; Otto, P.; Zimmerman, K.; Vidugiris, G.; Machleidt, T.; et al. Engineered luciferase reporter from a deep sea shrimp utilizing a novel imidazopyrazinone substrate. ACS Chem. Biol. 2012, 7, 1848–1857. [Google Scholar] [CrossRef]
- Shaner, N.C.; Campbell, R.E.; Steinbach, P.A.; Giepmans, B.N.G.; Palmer, A.E.; Tsien, R.Y. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat. Biotechnol. 2004, 22, 1567–1572. [Google Scholar] [CrossRef]
- Lodha, M.; Schulz-Raffelt, M.; Schroda, M. A new assay for promoter analysis in Chlamydomonas reveals roles for heat shock elements and the TATA box in HSP70A promoter-mediated activation of transgene expression. Eukaryot. Cell 2008, 7, 172–176. [Google Scholar] [CrossRef]
- Fages-Lartaud, M.; Tietze, L.; Elie, F.; Lale, R.; Hohmann-Marriott, M.F. mCherry contains a fluorescent protein isoform that interferes with its reporter function. Front. Bioeng. Biotechnol. 2022, 10, 892138. [Google Scholar] [CrossRef]
- Carroll, P.; Muwanguzi-Karugaba, J.; Melief, E.; Files, M.; Parish, T. Identification of the translational start site of codon-optimized mCherry in Mycobacterium tuberculosis. BMC Res. Notes 2014, 7, 366. [Google Scholar] [CrossRef]
- Eichler-Stahlberg, A.; Weisheit, W.; Ruecker, O.; Heitzer, M. Strategies to facilitate transgene expression in Chlamydomonas reinhardtii. Planta 2009, 229, 873–883. [Google Scholar] [CrossRef] [PubMed]
- Lumbreras, V.; Stevens, D.R.; Purton, S. Efficient foreign gene expression in Chlamydomonas reinhardtii mediated by an endogenous intron. Plant J. 1998, 14, 441–447. [Google Scholar] [CrossRef]
- Hinnebusch, A.G.; Ivanov, I.P.; Sonenberg, N. Translational control by 5′-untranslated regions of eukaryotic mRNAs. Science 2016, 352, 1413–1416. [Google Scholar] [CrossRef] [PubMed]
- Kozak, M. Point mutations define a sequence flanking the AUG initiator codon that modulates translation by eukaryotic ribosomes. Cell 1986, 44, 283–292. [Google Scholar] [CrossRef]
- Cross, F.R. Tying down loose ends in the Chlamydomonas genome: Functional significance of abundant upstream open reading frames. G3 (Bethesda) 2015, 6, 435–446. [Google Scholar] [CrossRef]
- Arribere, J.A.; Gilbert, W.V. Roles for transcript leaders in translation and mRNA decay revealed by transcript leader sequencing. Genome Res. 2013, 23, 977–987. [Google Scholar] [CrossRef]
- Kozak, M. Downstream secondary structure facilitates recognition of initiator codons by eukaryotic ribosomes. Proc. Natl. Acad. Sci. USA 1990, 87, 8301–8305. [Google Scholar] [CrossRef]
- Gruber, A.R.; Lorenz, R.; Bernhart, S.H.; Neuböck, R.; Hofacker, I.L. The Vienna RNA Websuite. Nucleic Acids Res. 2008, 36, W70–W74. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niemeyer, J.; Fischer, L.; Aylward, F.O.; Schroda, M. Analysis of Viral Promoters for Transgene Expression and of the Effect of 5′-UTRs on Alternative Translational Start Sites in Chlamydomonas. Genes 2023, 14, 948. https://doi.org/10.3390/genes14040948
Niemeyer J, Fischer L, Aylward FO, Schroda M. Analysis of Viral Promoters for Transgene Expression and of the Effect of 5′-UTRs on Alternative Translational Start Sites in Chlamydomonas. Genes. 2023; 14(4):948. https://doi.org/10.3390/genes14040948
Chicago/Turabian StyleNiemeyer, Justus, Laura Fischer, Frank O’Neill Aylward, and Michael Schroda. 2023. "Analysis of Viral Promoters for Transgene Expression and of the Effect of 5′-UTRs on Alternative Translational Start Sites in Chlamydomonas" Genes 14, no. 4: 948. https://doi.org/10.3390/genes14040948