

Genetic Screening of Targeted Region on the Chromosome 22q11.2 in Patients with Microtia and Congenital Heart Defect
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Participants
2.2. Targeted Region Capture Sequencing
2.3. Quality Control of Sequencing Data and Single Nucleotide Variant (SNV) Calling
2.4. Functional Annotation and Gene Burden Analysis
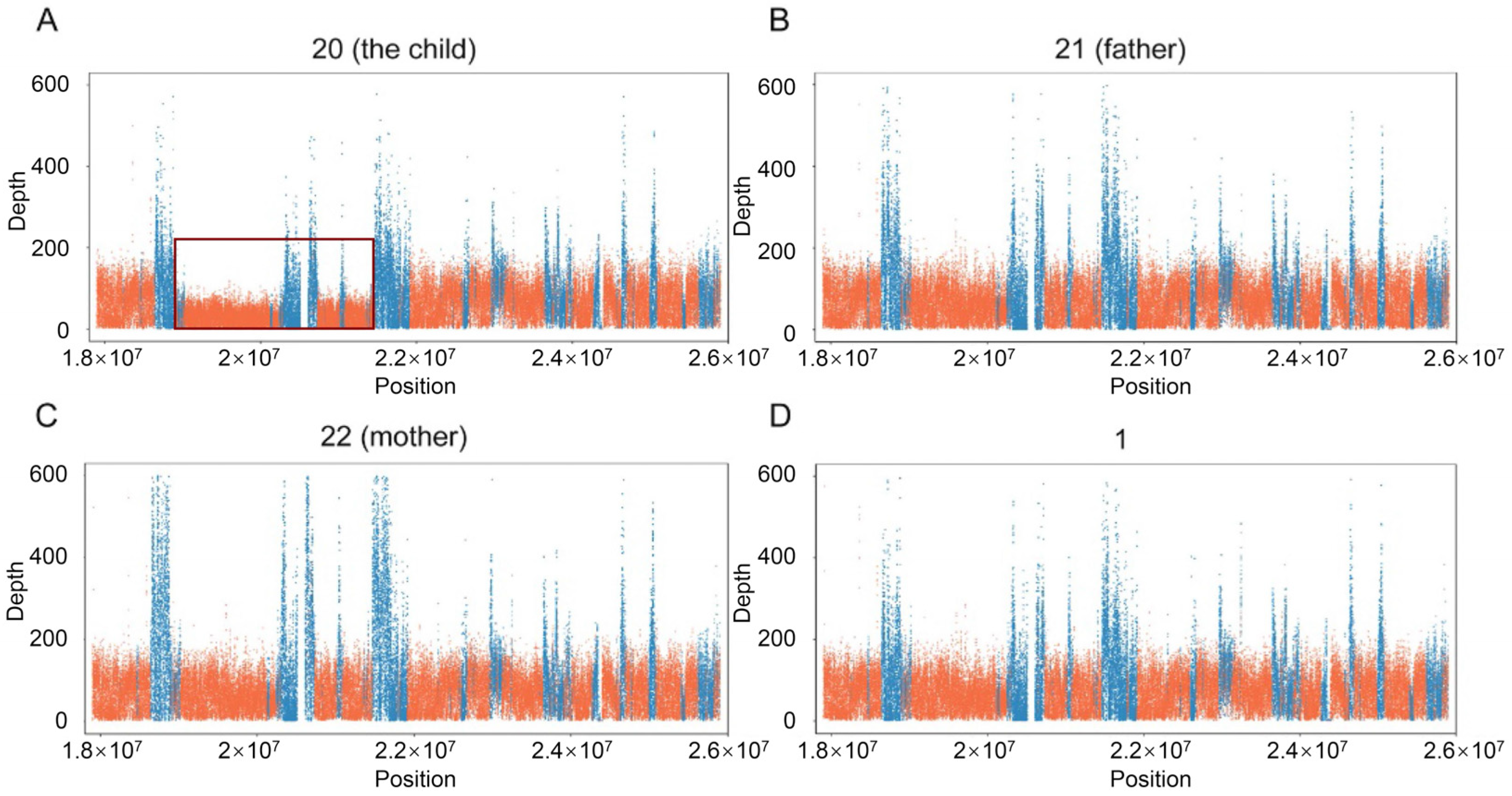
2.5. Copy Number Variant (CNV) Analysis and Validation by qRT-PCR
3. Results
3.1. Clinical Description
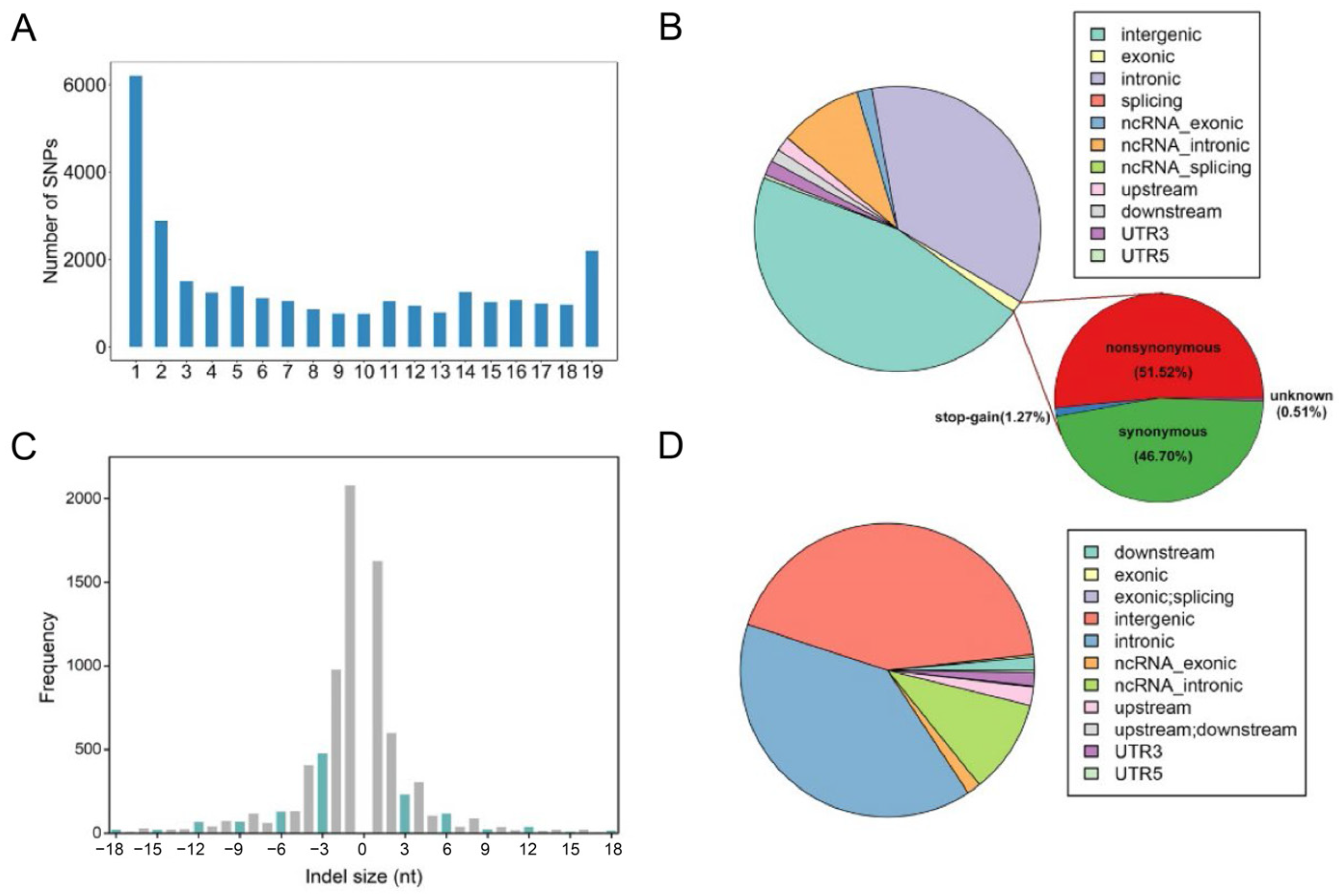
3.2. Functional Annotation of SNVs
3.3. Identification of Genes with Potential Functional Consequences
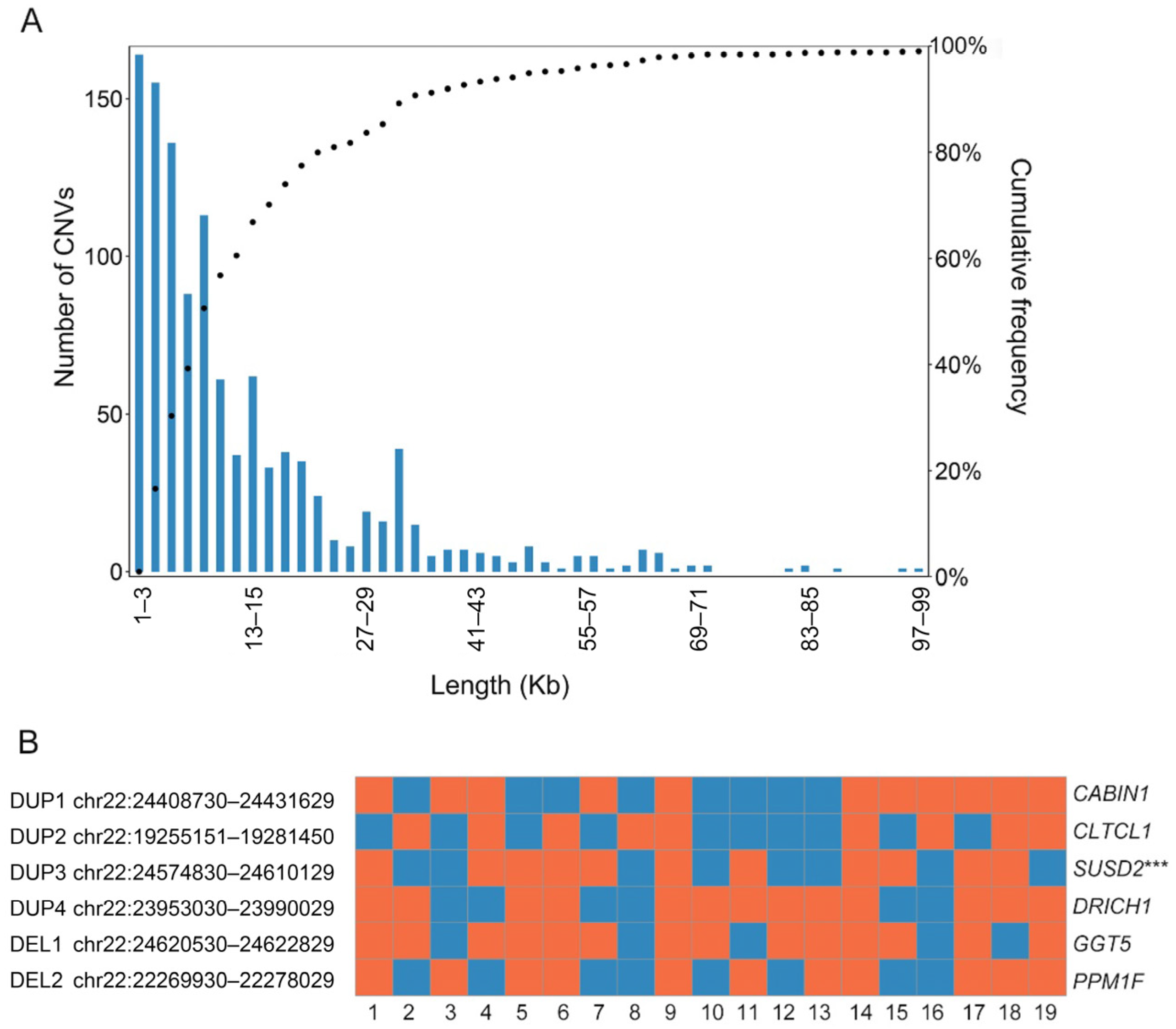
3.4. Microduplication Fragment May Associate with Microtia and CHD
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alasti, F.; Van Camp, G. Genetics of microtia and associated syndromes. J. Med. Genet. 2009, 46, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Suutarla, S.; Rautio, J.; Ritvanen, A.; Ala-Mello, S.; Jero, J.; Klockars, T. Microtia in Finland: Comparison of characteristics in different populations. Int. J. Pediatr. Otorhinolaryngol. 2007, 71, 1211–1217. [Google Scholar] [CrossRef]
- Chen, J.P.; Zhang, L.; Chen, G.; Song, X.M.; Zheng, X.Y. Capacity of monitoring system on birth defects during 1990s in China. Chin. J. Epidemiol. 2006, 27, 392. [Google Scholar]
- Luquetti, D.V.; Heike, C.L.; Hing, A.V.; Cunningham, M.L.; Cox, T.C. Microtia: Epidemiology and genetics†. Am. J. Med. Genet. A 2012, 158A, 124–139. [Google Scholar] [CrossRef] [Green Version]
- Wyse, R.K.; Al-Mahdawi, S.; Burn, J.; Blake, K. Congenital heart disease in CHARGE association. Pediatr. Cardiol. 1993, 14, 75–81. [Google Scholar] [CrossRef]
- Digilio, M.C.; Calzolari, F.; Capolino, R.; Toscano, A.; Sarkozy, A.; De Zorzi, A.; Dallapiccola, B.; Marino, B. Congenital heart defects in patients with oculo-auriculo-vertebral spectrum (Goldenhar syndrome). Am. J. Med. Genet. Part A 2008, 146, 1815–1819. [Google Scholar] [CrossRef]
- Zhang, Y.; Jiang, H.; Yang, Q.; He, L.; Yu, X.; Huang, X.; Wu, R.; Yang, M.; Li, C.; Pan, B. Microtia in a Chinese Specialty Clinic Population: Clinical Heterogeneity and Associated Congenital Anomalies. Plast. Reconstr. Surg. 2018, 142, 892e–903e. [Google Scholar] [CrossRef]
- Cao, T.; Chen, Q.; Wang, B.; Hu, J.; Zou, M.; Zhang, Q. Epidemiological research of microtia combined with congenital heart disease. J. Craniofacial Surg. 2021, 32, 920–924. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, T.; Kurahashi, H.; Saitta, S.; O’Hare, A.; Hu, P.; Roe, B.; Driscoll, D.; McDonald-McGinn, D.; Zackai, E.; Budarf, M.; et al. Chromosome 22-specific low copy repeats and the 22q11.2 deletion syndrome: Genomic organization and deletion endpoint analysis. Hum. Mol. Genet. 2000, 9, 489–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edelmann, L.; Pandita, R.K.; Spiteri, E.; Funke, B.; Goldberg, R.; Palanisamy, N.; Chaganti, R.S.K.; Magenis, E.; Shprintzen, R.J.; Morrow, B.E. A Common Molecular Basis for Rearrangement Disorders on Chromosome 22q11. Hum. Mol. Genet. 1999, 8, 1157. [Google Scholar] [CrossRef] [Green Version]
- McDonald-McGinn, D.M.; Sullivan, K.E.; Marino, B.; Philip, N.; Swillen, A.; Vorstman, J.A.; Zackai, E.H.; Emanuel, B.S.; Vermeesch, J.R.; Morrow, B.E. 22q11. 2 deletion syndrome. Nat. Rev. Dis. Prim. 2015, 1, 15071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Rocco, M.; Buocompagni, A.; Picco, P.; Vignola, S.; Borrone, C.; Gimelli, G. Spectrum of clinical features associated with interstitial chromosome 22q11 deletions. J. Med. Genet. 1998, 35, 346. [Google Scholar] [CrossRef] [Green Version]
- McDonald-McGinn, D.M.; Sullivan, K.E. Chromosome 22q11. 2 deletion syndrome (DiGeorge syndrome/velocardiofacial syndrome). Medicine 2011, 90, 1–18. [Google Scholar] [CrossRef] [PubMed]
- White, L.K.; Crowley, T.B.; Finucane, B.; McClellan, E.J.; Donoghue, S.; Garcia-Minaur, S.; Repetto, G.M.; Fischer, M.; Jacquemont, S.; Gur, R.E.; et al. Gathering the Stakeholder’s Perspective: Experiences and Opportunities in Rare Genetic Disease Research. Genes 2023, 14, 169. [Google Scholar]
- Tan, T.Y.; Collins, A.; James, P.A.; McGillivray, G.; Stark, Z.; Gordon, C.T.; Leventer, R.J.; Pope, K.; Forbes, R.; Crolla, J.A.; et al. Phenotypic variability of distal 22q11. 2 copy number abnormalities. Am. J. Med. Genet. A. 2011, 155, 1623–1633. [Google Scholar] [CrossRef] [PubMed]
- Blagowidow, N.; Nowakowska, B.; Schindewolf, E.; Grati, F.R.; Putotto, C.; Breckpot, J.; Swillen, A.; Crowley, T.B.; Loo, J.C.Y.; Lairson, L.A.; et al. Prenatal sreening and diagnostic considerations for 22q11.2 microdeletions. Genes 2023, 14, 160. [Google Scholar] [CrossRef]
- Hunter, A.; Frias, J.L.; Gillessen-Kaesbach, G.; Hughes, H.; Jones, K.L.; Wilson, L. Elements of morphology: Standard terminology for the ear. Am. J. Med. Genet. Part A 2009, 149a, 40–60. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 12 November 2017).
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J. From FastQ data to high-confidence variant calls: The genome analysis toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.11–11.10.33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sudmant, P.H.; Rausch, T.; Gardner, E.J.; Handsaker, R.E.; Abyzov, A.; Huddleston, J.; Zhang, Y.; Ye, K.; Jun, G.; Fritz, M.H.-Y. An integrated map of structural variation in 2,504 human genomes. Nature 2015, 526, 75. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Jian, X.; Boerwinkle, E. dbNSFP v2. 0: A database of human non-synonymous SNVs and their functional predictions and annotations. Hum. Mutat. 2013, 34, E2393–E2402. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Gamazon, E.R.; Zhang, X.; Konkashbaev, A.; Liu, C.; Szilágyi, K.L.; Dolan, M.E.; Cox, N.J. SCAN database: Facilitating integrative analyses of cytosine modification and expression QTL. Database 2015, 2015, bav025. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Abyzov, A.; Urban, A.E.; Snyder, M.; Gerstein, M. CNVnator: An approach to discover, genotype and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 2011, 21, 974–984. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pruitt, K.D.; Tatusova, T.; Maglott, D.R. NCBI Reference Sequence (RefSeq): A curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2005, 33, D501–D504. [Google Scholar] [CrossRef] [Green Version]
- Forrester, M.B.; Merz, R.D. Descriptive epidemiology of anotia and microtia, Hawaii, 1986–2002. Congenit. Anom. 2005, 45, 119–124. [Google Scholar] [CrossRef]
- Okajima, H.; Takeichi, Y.; Umeda, K.; Baba, S. Clinical analysis of 592 patients with microtia. Acta Oto-Laryngol. Suppl. 1996, 525, 18–24. [Google Scholar]
- Shaw, G.M.; Carmichael, S.L.; Kaidarova, Z.; Harris, J.A. Epidemiologic characteristics of anotia and microtia in California, 1989–1997. Birth Defects Res. Part A Clin. Mol. Teratol. 2004, 70, 472–475. [Google Scholar] [CrossRef] [PubMed]
- van der Linde, D.; Konings, E.E.M.; Slager, M.A.; Witsenburg, M.; Helbing, W.A.; Takkenberg, J.J.M.; Roos-Hesselink, J.W. Birth Prevalence of Congenital Heart Disease Worldwide. A Syst. Rev. Meta-Anal. 2011, 58, 2241–2247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balcı, S.; Engiz, Ö. Goldenhar syndrome phenotypes and 22q11 deletion. Am. J. Med. Genet. Part A 2011, 155, 458. [Google Scholar] [CrossRef] [PubMed]
- Jerome, L.A.; Papaioannou, V.E. DiGeorge syndrome phenotype in mice mutant for the T-box gene, Tbx1. Nat. Genet. 2001, 27, 286. [Google Scholar] [CrossRef]
- Papangeli, I.; Scambler, P. The 22q11 deletion: DiGeorge and velocardiofacial syndromes and the role of TBX1. Wiley Interdiscip. Rev. Dev. Biol. 2013, 2, 393–403. [Google Scholar] [CrossRef]
- Yagi, H.; Furutani, Y.; Hamada, H.; Sasaki, T.; Asakawa, S.; Minoshima, S.; Ichida, F.; Joo, K.; Kimura, M.; Imamura, S.-I.; et al. Role of TBX1 in human del22q11.2 syndrome. Lancet 2003, 362, 1366–1373. [Google Scholar] [CrossRef]
- Calmont, A.; Ivins, S.; Van Bueren, K.L.; Papangeli, I.; Kyriakopoulou, V.; Andrews, W.D.; Martin, J.F.; Moon, A.M.; Illingworth, E.A.; Basson, M.A. Tbx1 controls cardiac neural crest cell migration during arch artery development by regulating Gbx2 expression in the pharyngeal ectoderm. Development 2009, 136, 3173–3183. [Google Scholar] [CrossRef] [Green Version]
- Vitelli, F.; Morishima, M.; Taddei, I.; Lindsay, E.A.; Baldini, A. Tbx1 mutation causes multiple cardiovascular defects and disrupts neural crest and cranial nerve migratory pathways. Hum. Mol. Genet. 2002, 11, 915–922. [Google Scholar] [CrossRef] [Green Version]
- Merscher, S.; Funke, B.; Epstein, J.A.; Heyer, J.; Puech, A.; Lu, M.M.; Xavier, R.J.; Demay, M.B.; Russell, R.G.; Factor, S. TBX1 is responsible for cardiovascular defects in velo-cardio-facial/DiGeorge syndrome. Cell 2001, 104, 619–629. [Google Scholar] [CrossRef] [Green Version]
- Lindsay, E.A.; Vitelli, F.; Su, H.; Morishima, M.; Huynh, T.; Pramparo, T.; Jurecic, V.; Ogunrinu, G.; Sutherland, H.F.; Scambler, P.J.; et al. Tbx1 haploinsufficiency in the DiGeorge syndrome region causes aortic arch defects in mice. Nature 2001, 410, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Medvid, R.; Melton, C.; Jaenisch, R.; Blelloch, R. DGCR8 is essential for microRNA biogenesis and silencing of embryonic stem cell self-renewal. Nat. Genet. 2007, 39, 380. [Google Scholar] [CrossRef] [PubMed]
- Stark, K.L.; Xu, B.; Bagchi, A.; Lai, W.-S.; Liu, H.; Hsu, R.; Wan, X.; Pavlidis, P.; Mills, A.A.; Karayiorgou, M. Altered brain microRNA biogenesis contributes to phenotypic deficits in a 22q11-deletion mouse model. Nat. Genet. 2008, 40, 751. [Google Scholar] [CrossRef]
- Smith, T.; Rajakaruna, C.; Caputo, M.; Emanueli, C. MicroRNAs in congenital heart disease. Ann. Transl. Med. 2015, 3, 333. [Google Scholar]
- Chapnik, E.; Sasson, V.; Blelloch, R.; Hornstein, E. Dgcr8 controls neural crest cells survival in cardiovascular development. Dev. Biol. 2012, 362, 50–56. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Wang, X.; Wang, P.; Li, T.; Hu, F.; Liu, Q.; Yang, F.; Wang, J.; Xu, T.; Han, W. SUSD2 is frequently downregulated and functions as a tumor suppressor in RCC and lung cancer. Tumor Biol. 2016, 37, 9919–9930. [Google Scholar] [CrossRef]
- Watson, A.P.; Evans, R.L.; Egland, K.A. Multiple functions of sushi domain containing 2 (SUSD2) in breast tumorigenesis. Mol. Cancer Res. 2013, 11, 74–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nadjar, Y.; Triller, A.; Bessereau, J.-L.; Dumoulin, A. The Susd2 protein regulates neurite growth and excitatory synaptic density in hippocampal cultures. Mol. Cell. Neurosci. 2015, 65, 82–91. [Google Scholar] [CrossRef]
- Shi, X.; Huang, T.; Wang, J.; Liang, Y.; Gu, C.; Xu, Y.; Sun, J.; Lu, Y.; Sun, K.; Chen, S.; et al. Next-generation sequencing identifies novel genes with rare variants in total anomalous pulmonary venous connection. EBioMedicine 2018, 38, 217–227. [Google Scholar] [CrossRef] [Green Version]
- Bard, F.; Casano, L.; Mallabiabarrena, A.; Wallace, E.; Saito, K.; Kitayama, H.; Guizzunti, G.; Hu, Y.; Wendler, F.; DasGupta, R.; et al. Functional genomics reveals genes involved in protein secretion and Golgi organization. Nature 2006, 439, 604–607. [Google Scholar] [CrossRef] [PubMed]
- Kremer, L.S.; Distelmaier, F.; Alhaddad, B.; Hempel, M.; Iuso, A.; Küpper, C.; Mühlhausen, C.; Kovacs-Nagy, R.; Satanovskij, R.; Graf, E.; et al. Bi-allelic Truncating Mutations in TANGO2 Cause Infancy-Onset Recurrent Metabolic Crises with Encephalocardiomyopathy. Am. J. Hum. Genet. 2016, 98, 358–362. [Google Scholar] [CrossRef] [Green Version]
- Lalani, S.R.; Liu, P.; Rosenfeld, J.A.; Watkin, L.B.; Chiang, T.; Leduc, M.S.; Zhu, W.; Ding, Y.; Pan, S.; Vetrini, F.; et al. Recurrent Muscle Weakness with Rhabdomyolysis, Metabolic Crises, and Cardiac Arrhythmia Due to Bi-allelic TANGO2 Mutations. Am. J. Hum. Genet. 2016, 98, 347–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dines, J.N.; Golden-Grant, K.; LaCroix, A.; Muir, A.M.; Cintrón, D.L.; McWalter, K.; Cho, M.T.; Sun, A.; Merritt, J.L.; Thies, J. TANGO2: Expanding the clinical phenotype and spectrum of pathogenic variants. Genet. Med. 2019, 21, 601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motahari, Z.; Moody, S.A.; Maynard, T.M.; LaMantia, A.-S. In the line-up: Deleted genes associated with DiGeorge/22q11.2 deletion syndrome: Are they all suspects? J. Neurodev. Disord. 2019, 11, 7. [Google Scholar] [CrossRef] [Green Version]
- Acuna, C.; Liu, X.; Gonzalez, A.; Südhof Thomas, C. RIM-BPs Mediate Tight Coupling of Action Potentials to Ca2+-Triggered Neurotransmitter Release. Neuron 2015, 87, 1234–1247. [Google Scholar] [CrossRef] [Green Version]
- Gao, T.; Zhang, Z.; Yang, Y.; Zhang, H.; Li, N.; Liu, B. Impact of RIM-BPs in neuronal vesicles release. Brain Res. Bull. 2021, 170, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Jungerius, B.J.; Hoogendoorn, M.L.C.; Bakker, S.C.; van’t Slot, R.; Bardoel, A.F.; Ophoff, R.A.; Wijmenga, C.; Kahn, R.S.; Sinke, R.J. An association screen of myelin-related genes implicates the chromosome 22q11 PIK4CA gene in schizophrenia. Mol. Psychiatry 2008, 13, 1060–1068. [Google Scholar] [CrossRef] [Green Version]
- Pagnamenta, A.T.; Howard, M.F.; Wisniewski, E.; Popitsch, N.; Knight, S.J.; Keays, D.A.; Quaghebeur, G.; Cox, H.; Cox, P.; Balla, T.; et al. Germline recessive mutations in PI4KA are associated with perisylvian polymicrogyria, cerebellar hypoplasia and arthrogryposis. Hum. Mol. Genet. 2015, 24, 3732–3741. [Google Scholar] [CrossRef] [Green Version]
- Ye, Y.; Meyer, H.H.; Rapoport, T.A. Function of the p97–Ufd1–Npl4 complex in retrotranslocation from the ER to the cytosol: Dual recognition of nonubiquitinated polypeptide segments and polyubiquitin chains. J. Cell Biol. 2003, 162, 71–84. [Google Scholar] [CrossRef]
- Ye, Y.; Meyer, H.H.; Rapoport, T.A. The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature 2001, 414, 652–656. [Google Scholar] [CrossRef]
- Bays, N.W.; Hampton, R.Y. Cdc48–Ufd1–Npl4: Stuck in the middle with Ub. Curr. Biol. 2002, 12, R366–R371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novelli, G.; Mari, A.; Amati, F.; Colosimo, A.; Sangiuolo, F.; Bengala, M.; Conti, E.; Ratti, A.; Bordoni, R.; Pizzuti, A. Structure and expression of the human ubiquitin fusion–degradation gene (UFD1L). Biochim. Biophys. Acta (BBA)-Gene Struct. Expr. 1998, 1396, 158–162. [Google Scholar] [CrossRef]
- Yamagishi, H. A Molecular Pathway Revealing a Genetic Basis for Human Cardiac and Craniofacial Defects. Science 1999, 283, 1158–1161. [Google Scholar] [CrossRef] [Green Version]
- Rizzu, P.; Lindsay, E.A.; Taylor, C.; O’Donnell, H.; Levy, A.; Scambler, P.; Baldini, A. Cloning and comparative mapping of a gene from the commonly deleted region of DiGeorge and Velocardiofacial syndromes conserved in C. elegans. Mamm. Genome 1996, 7, 639–643. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, S.A.; Hanke, T.; Schlueter, C.; Bullerdiek, J.; Sievers, H.-H. Ubiquitin fusion degradation 1–like gene dysregulation in bicuspid aortic valve. J. Thorac. Cardiovasc. Surg. 2005, 130, 1531–1536. [Google Scholar] [CrossRef] [Green Version]
- Zarrei, M.; MacDonald, J.R.; Merico, D.; Scherer, S.W. A copy number variation map of the human genome. Nat. Rev. Genet. 2015, 16, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Sharp, A.J.; Locke, D.P.; McGrath, S.D.; Cheng, Z.; Bailey, J.A.; Vallente, R.U.; Pertz, L.M.; Clark, R.A.; Schwartz, S.; Segraves, R.; et al. Segmental duplications and copy-number variation in the human genome. Am. J. Hum. Genet. 2005, 77, 78–88. [Google Scholar] [CrossRef] [Green Version]
- Glessner, J.T.; Wang, K.; Cai, G.; Korvatska, O.; Kim, C.E.; Wood, S.; Zhang, H.; Estes, A.; Brune, C.W.; Bradfield, J.P.; et al. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature 2009, 459, 569–573. [Google Scholar] [CrossRef] [Green Version]
- Diskin, S.J.; Hou, C.; Glessner, J.T.; Attiyeh, E.F.; Laudenslager, M.; Bosse, K.; Cole, K.; Mosse, Y.P.; Wood, A.; Lynch, J.E.; et al. Copy number variation at 1q21.1 associated with neuroblastoma. Nature 2009, 459, 987–991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bochukova, E.G.; Huang, N.; Keogh, J.; Henning, E.; Purmann, C.; Blaszczyk, K.; Saeed, S.; Hamilton-Shield, J.; Clayton-Smith, J.; O’Rahilly, S.; et al. Large, rare chromosomal deletions associated with severe early-onset obesity. Nature 2010, 463, 666–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moutsianas, L.; Agarwala, V.; Fuchsberger, C.; Flannick, J.; Rivas, M.A.; Gaulton, K.J.; Albers, P.K.; Go, T.D.C.; McVean, G.; Boehnke, M.; et al. The power of gene-based rare variant methods to detect disease-associated variation and test hypotheses about complex disease. PLoS Genet. 2015, 11, e1005165. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Sex | Age | Ear Anomalies | Cardiac Anomalies | Other Anomalies |
|---|---|---|---|---|---|
| 1 | M | 8 | B; degree I | VSD | |
| 2 | M | 6 | R; degree I | VSD | |
| 3 | F | 20 | R; degree I | TOF | |
| 4 | M | 13 | L; degree I | VSD; TI; PI | |
| 5 | M | 25 | L; degree I | VSD | |
| 6 | M | 9 | L; degree I | TOF | |
| 7 | M | 6 | R; degree II | PDA | |
| 8 | M | 12 | R; degree II | VSD; PH | |
| 9 | M | 11 | R; degree II | VSD | cleft palate |
| 10 | M | 8 | R; degree II | TOF | |
| 11 | M | 7 | R; degree II | PFO | bilateral preauricular fistula |
| 12 | F | 13 | R; degree II | ASD | |
| 13 | M | 10 | R; degree II | VSD | |
| 14 | M | 18 | L; degree II | VSD | |
| 15 | M | 15 | L; degree II | ASD | |
| 16 | F | 6 | L; degree II | VSD | |
| 17 | M | 7 | L; degree II | TOF | |
| 18 | F | 35 | R; degree III | ASD | facial cleft; hemifacial dysplasia |
| 19 | F | 8 | R; degree III | ASD | hemifacial dysplasia; facial transversal cleft; left eye cyst |
| 20 1 | M | 7 | B; degree I | ASD; PDA | polydactyly; epilepsy |
| Gene | Counts (CHB) 1 | Counts (Cases) | Frequency Ratio | p Value |
|---|---|---|---|---|
| SUSD2 | 5 | 20 | 21.68 | <0.0001 |
| COMT | 9 | 2 | 1.20 | <0.0001 |
| GGT2 | 16 | 22 | 7.45 | <0.0001 |
| TOP3B | 16 | 4 | 1.36 | <0.0001 |
| SCARF2 | 29 | 6 | 1.12 | <0.0001 |
| PIWIL3 | 1 | 3 | 16.26 | 0.0001 |
| BCR | 2 | 2 | 5.42 | 0.0002 |
| TBX1 | 38 | 8 | 1.14 | 0.0293 |
| TANGO2 | 1 | 1 | 5.42 | 0.0350 |
| CLTCL1 | 116 | 25 | 1.17 | 0.0430 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, C.; Yang, Y.; Pan, B.; Wei, H.; Ju, J.; Si, N.; Xu, Q. Genetic Screening of Targeted Region on the Chromosome 22q11.2 in Patients with Microtia and Congenital Heart Defect. Genes 2023, 14, 879. https://doi.org/10.3390/genes14040879
Zhu C, Yang Y, Pan B, Wei H, Ju J, Si N, Xu Q. Genetic Screening of Targeted Region on the Chromosome 22q11.2 in Patients with Microtia and Congenital Heart Defect. Genes. 2023; 14(4):879. https://doi.org/10.3390/genes14040879
Chicago/Turabian StyleZhu, Caiyun, Yang Yang, Bo Pan, Hui Wei, Jiahang Ju, Nuo Si, and Qi Xu. 2023. "Genetic Screening of Targeted Region on the Chromosome 22q11.2 in Patients with Microtia and Congenital Heart Defect" Genes 14, no. 4: 879. https://doi.org/10.3390/genes14040879