
Whole Genome Sequencing Provides Information on the Genomic Architecture and Diversity of Cultivated Gilthead Seabream (Sparus aurata) Broodstock Nuclei
 , , ,
, , ,  , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Gilthead Seabream Populations and Genomic Datasets
2.2. Sequencing, Alignment, and Variant Calling
2.3. Population Genomic Analyses and Genome Annotation
3. Results and Discussion
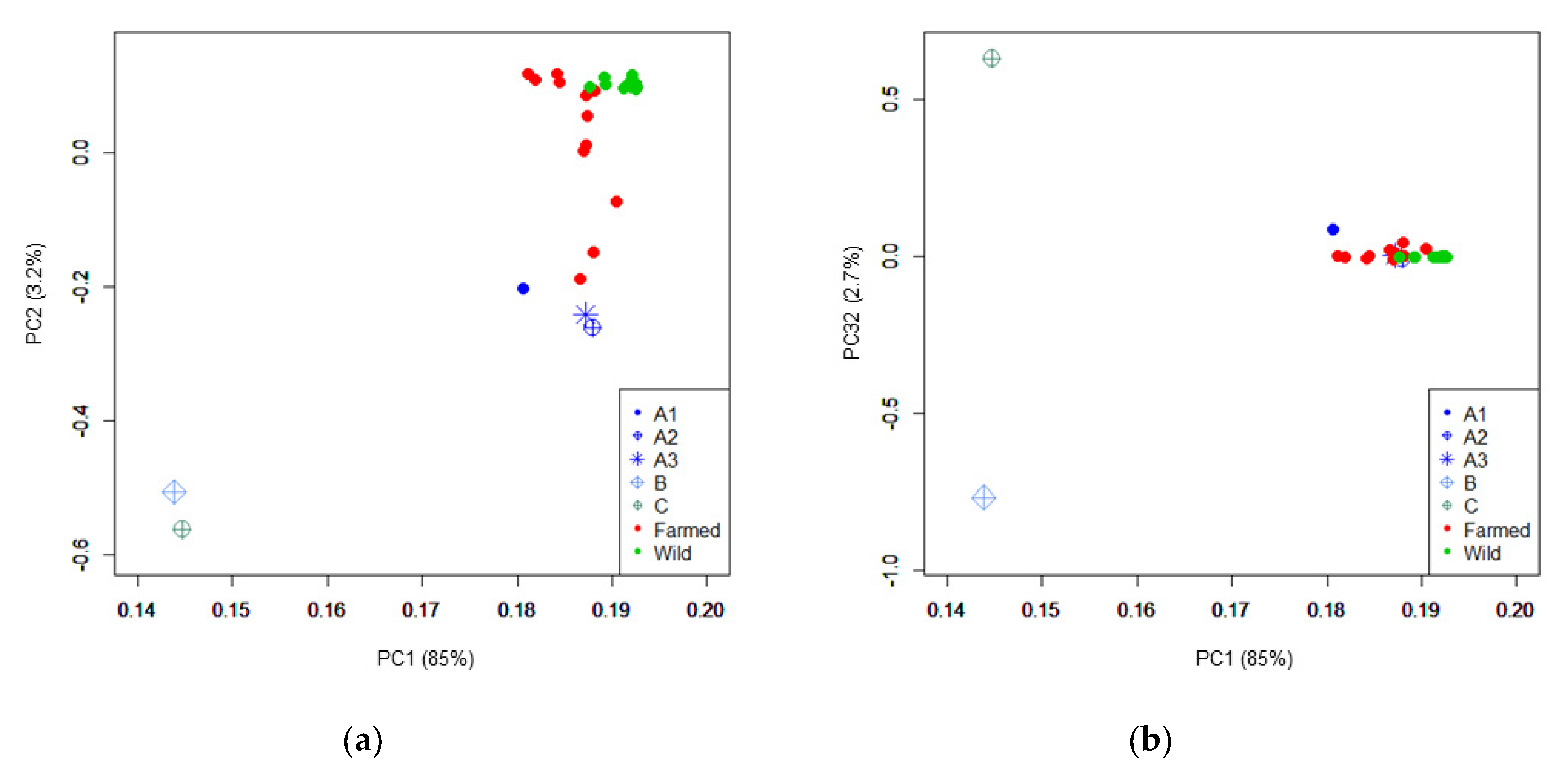
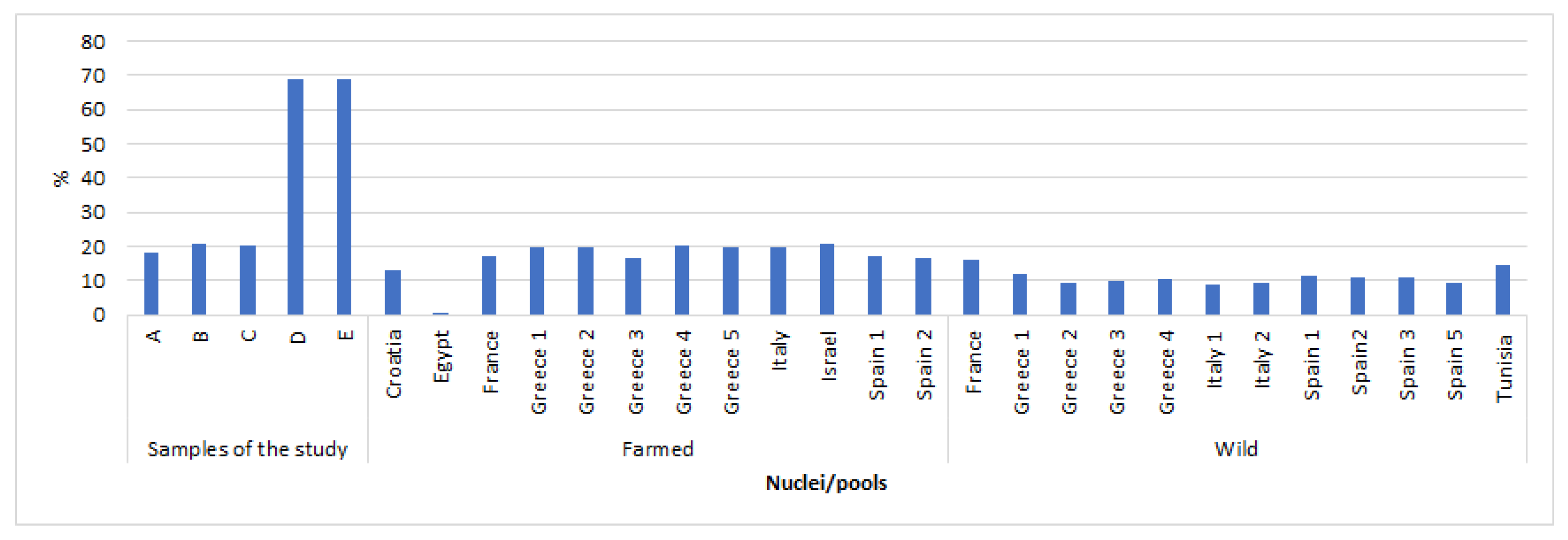
3.1. Population Genomic Parameters of Different Gilthead Seabream Stocks
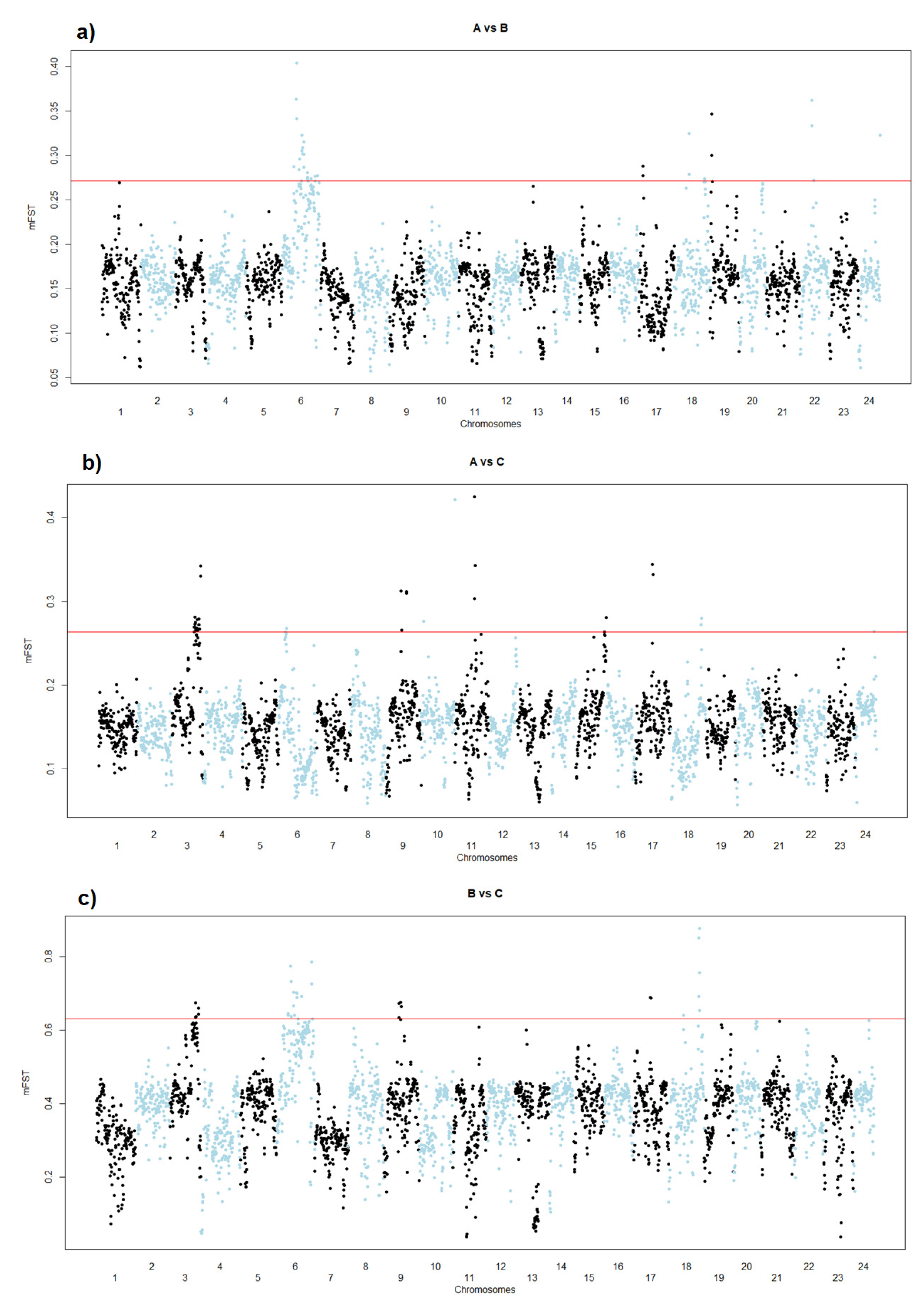
3.2. Genome-Wide Window-Based FST Analyses
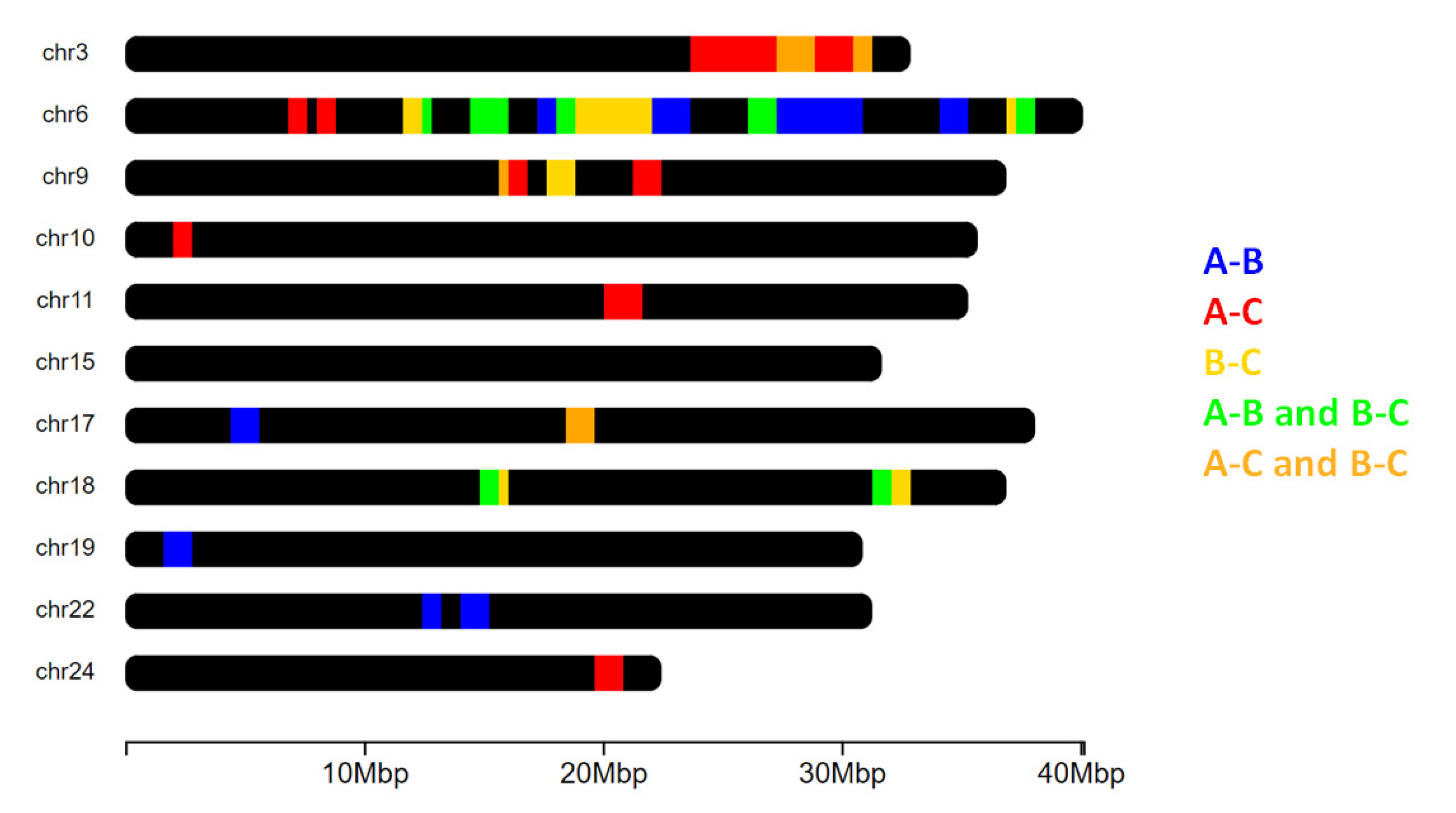
3.3. Functional Annotation of Outliers FST Window Regions
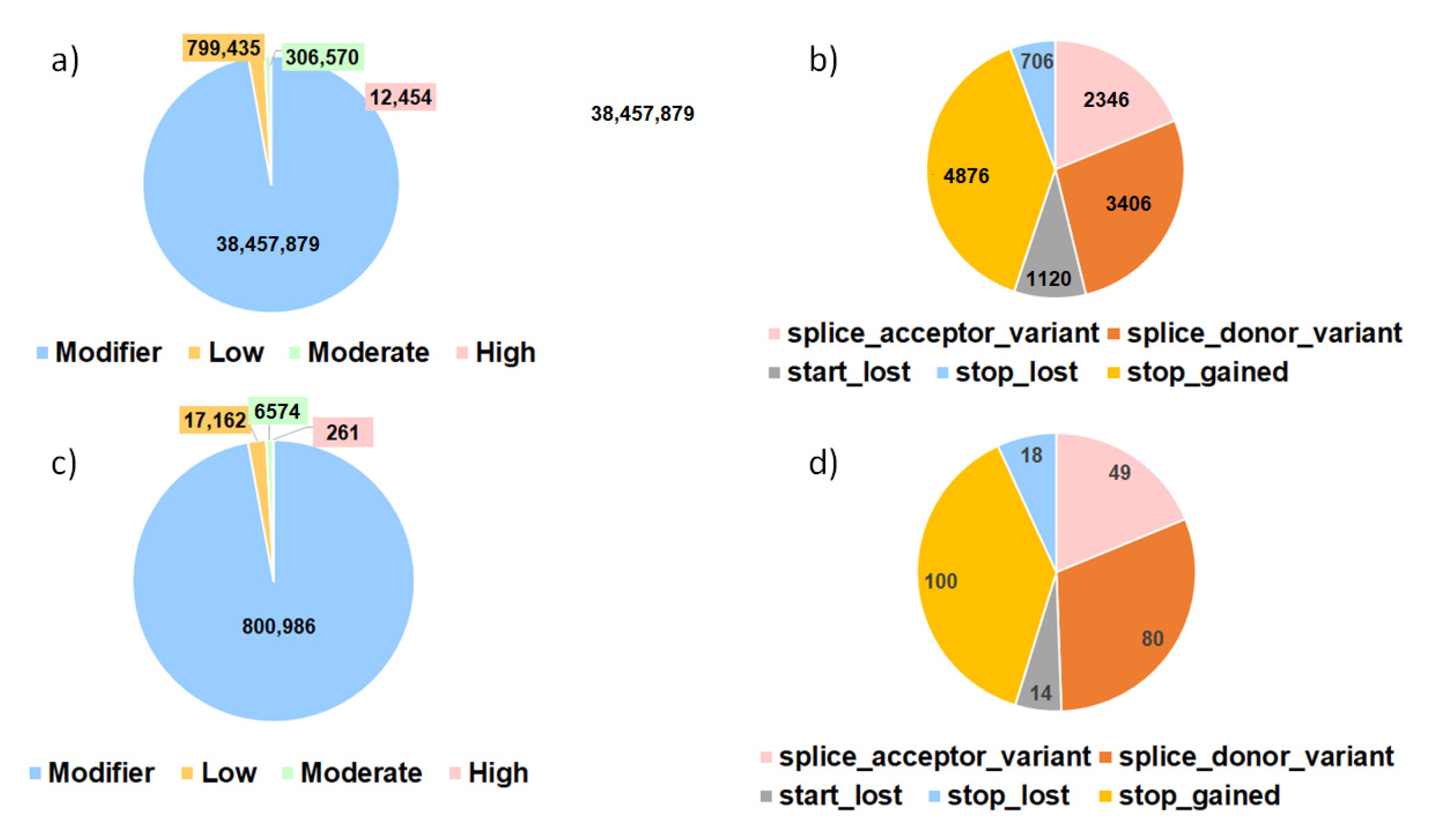
3.3.1. Impacts of SNPs
3.3.2. Overrepresentation of Biological Processes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- EUROSTAT. Production from Aquaculture Excluding Hatcheries and Nurseries (fish_AQ2A Datasheet). 2020. Available online: https://ec.europa.eu/eurostat/databrowser/view/FISH_AQ2A__custom_4842525/default/table?lang=en (accessed on 5 February 2023).
- Manchado, M.; Planas, J.V.; Cousin, X.; Rebordinos, L.; Claros, M.G. Surrent status in other finfish species: Description of current genomic resources for the Gilthead seabream (Sparus aurata) and soles (Solea senegalensis and Solea solea). In Genomics in Aquaculture; Academic Press: Cambridge, MA, USA, 2016; pp. 195–221. [Google Scholar]
- Moretti, A.; Fernandez-Criado, M.P.; Cittolin, G. Ruggero Guidastri Manual on Hatchery Production of Seabass and Gilthead Seabream; Food & Agriculture Organization: Quebec City, QC, Canada, 1999; Volume 1. [Google Scholar]
- Ferosekhan, S.; Turkmen, S.; Pérez-García, C.; Xu, H.; Gómez, A.; Shamna, N.; Afonso, J.M.; Rosenlund, G.; Fontanillas, R.; Gracia, A.; et al. Influence of genetic selection for growth and broodstock diet N-3 LC-PUFA levels on reproductive performance of Gilthead seabream, Sparus aurata. Animals 2021, 11, 519. [Google Scholar] [CrossRef] [PubMed]
- Boudry, P.; Allal, F.; Aslam, M.L.; Bargelloni, L.; Bean, T.P.; Brard-Fudulea, S.; Brieuc, M.S.O.; Calboli, F.C.F.; Gilbey, J.; Haffray, P.; et al. Current status and potential of genomic selection to improve selective breeding in the main aquaculture species of international council for the exploration of the sea (ICES) member countries. Aquac. Rep. 2021, 20, 100700. [Google Scholar] [CrossRef]
- Pauletto, M.; Manousaki, T.; Ferraresso, S.; Babbucci, M.; Tsakogiannis, A.; Louro, B.; Vitulo, N.; Quoc, V.H.; Carraro, R.; Bertotto, D.; et al. Genomic analysis of sparus aurata reveals the evolutionary dynamics of sex-biased genes in a sequential hermaphrodite fish. Commun. Biol. 2018, 1, 119. [Google Scholar] [CrossRef] [PubMed]
- Peñaloza, C.; Manousaki, T.; Franch, R.; Tsakogiannis, A.; Sonesson, A.K.; Aslam, M.L.; Allal, F.; Bargelloni, L.; Houston, R.D.; Tsigenopoulos, C.S. Development and testing of a combined species snp array for the european seabass (Dicentrarchus labrax) and Gilthead seabream (Sparus aurata). Genomics 2021, 113, 2096–2107. [Google Scholar] [CrossRef]
- Besson, M.; Rombout, N.; Salou, G.; Vergnet, A.; Cariou, S.; Bruant, J.S.; Izquierdo, M.; Bestin, A.; Clota, F.; Haffray, P.; et al. Potential for genomic selection on feed efficiency in Gilthead sea bream (Sparus aurata), based on individual feed conversion ratio, carcass and lipid traits. Aquac. Rep. 2022, 24, 101132. [Google Scholar] [CrossRef]
- Palaiokostas, C.; Ferraresso, S.; Franch, R.; Houston, R.D.; Bargelloni, L. Genomic prediction of resistance to pasteurellosis in Gilthead sea bream (Sparus aurata) using 2B-RAD sequencing. G3 Genes Genomes Genet. 2016, 6, 3693–3700. [Google Scholar] [CrossRef] [Green Version]
- Aslam, M.L.; Carraro, R.; Bestin, A.; Cariou, S.; Sonesson, A.K.; Bruant, J.S.; Haffray, P.; Bargelloni, L.; Meuwissen, T.H.E. Genetics of resistance to photobacteriosis in Gilthead sea bream (Sparus aurata) using 2b-RAD sequencing. BMC Genet. 2018, 19, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griot, R.; Allal, F.; Phocas, F.; Brard-Fudulea, S.; Morvezen, R.; Haffray, P.; François, Y.; Morin, T.; Bestin, A.; Bruant, J.S.; et al. Optimization of genomic selection to improve disease resistance in two marine fishes, the european Sea bass (Dicentrarchus labrax) and the Gilthead sea bream (Sparus aurata). Front. Genet. 2021, 12, 754416. [Google Scholar] [CrossRef]
- Bertolini, F.; Ribani, A.; Capoccioni, F.; Buttazzoni, L.; Utzeri, V.J.; Bovo, S.; Schiavo, G.; Caggiano, M.; Rothschild, M.F.; Fontanesi, L. A Comparative Whole Genome Sequencing analysis identified a candidate locus for lack of operculum in cultivated Gilthead seabream (Sparus aurata). Anim. Genet. 2021, 52, 365–370. [Google Scholar] [CrossRef]
- Bertolini, F.; Ribani, A.; Capoccioni, F.; Buttazzoni, L.; Utzeri, V.J.; Bovo, S.; Schiavo, G.; Caggiano, M.; Fontanesi, L.; Rothschild, M.F. Identification of a major locus determining a pigmentation defect in cultivated Gilthead seabream (Sparus aurata). Anim. Genet. 2020, 51, 319–323. [Google Scholar] [CrossRef]
- López, M.E.; Neira, R.; Yáñez, J.M. Applications in the search for genomic selection signatures in fish. Front. Genet. 2014, 5, 458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertolini, F.; Geraci, C.; Schiavo, G.; Sardina, M.T.; Chiofalo, V.; Fontanesi, L. Whole Genome Semiconductor Based Sequencing of farmed european Sea bass (Dicentrarchus labrax) mediterranean genetic stocks using a dna pooling approach. Mar. Genom. 2016, 28, 63–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, V.; Dettleff, P.; Lopez, P.; Fernandez, G.; Jedlicki, A.; Yañez, J.M.; Davidson, W.S. Assessing footprints of selection in commercial atlantic salmon populations using microsatellite data. Anim. Genet. 2013, 44, 223–226. [Google Scholar] [CrossRef] [PubMed]
- de Innocentiis, S.; Lesti, A.; Livi, S.; Rossi, A.R.; Crosetti, D.; Sola, L. Microsatellite markers reveal population structure in Gilthead sea bream Sparus aurata from the atlantic ocean and mediterranean sea. Fish. Sci. 2004, 70, 852–859. [Google Scholar] [CrossRef]
- Loukovitis, D.; Sarropoulou, E.; Vogiatzi, E.; Tsigenopoulos, C.S.; Kotoulas, G.; Magoulas, A.; Chatziplis, D. Genetic variation in farmed populations of the Gilthead sea bream Sparus aurata in Greece using microsatellite DNA markers. Aquac. Res. 2012, 43, 239–246. [Google Scholar] [CrossRef]
- Coscia, I.; Vogiatzi, E.; Kotoulas, G.; Tsigenopoulos, C.S.; Mariani, S. Exploring neutral and adaptive processes in expanding populations of Gilthead sea bream, Sparus aurata, in the north-east atlantic. Heredity 2012, 108, 537–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maroso, F.; Gkagkavouzis, K.; de Innocentiis, S.; Hillen, J.; do Prado, F.; Karaiskou, N.; Taggart, J.B.; Carr, A.; Nielsen, E.; Triantafyllidis, A.; et al. Genome-Wide analysis clarifies the population genetic structure of wild Gilthead sea bream (Sparus aurata). PLoS ONE 2021, 16, e0236230. [Google Scholar] [CrossRef]
- Segvic-Bubic, T.; Lepen, I.; Trumbic, Z.; Ljubkovic, J.; Sutlovic, D.; Matic-Skoko, S.; Grubisic, L.; Glamuzina, B.; Mladineo, I. Population genetic structure of reared and wild Gilthead sea bream (Sparus aurata) in the adriatic sea inferred with microsatellite loci. Aquaculture 2011, 318, 309–315. [Google Scholar] [CrossRef]
- Franchini, P.; Sola, L.; Crosetti, D.; Milana, V.; Rossi, A.R. Low Levels of population genetic structure in the Gilthead sea bream, sparus aurata, along the coast of Italy. ICES J. Mar. Sci. 2012, 69, 41–50. [Google Scholar] [CrossRef] [Green Version]
- Žužul, I.; Šegvić-Bubić, T.; Talijančić, I.; Džoić, T.; Lepen Pleić, I.; Beg Paklar, G.; Ivatek-Šahdan, S.; Katavić, I.; Grubišić, L. Spatial connectivity pattern of expanding Gilthead seabream populations and its interactions with aquaculture sites: A combined population genetic and physical modelling approach. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Guinand, B.; Chauvel, C.; Lechene, M.; Tournois, J.; Tsigenopoulos, C.S.; Darnaude, A.M.; McKenzie, D.J.; Gagnaire, P.A. Candidate gene variation in Gilthead sea bream reveals complex spatiotemporal selection patterns between marine and lagoon habitats. Mar. Ecol. Prog. Ser. 2016, 558, 115–127. [Google Scholar] [CrossRef] [Green Version]
- Gkagkavouzis, K.; Papakostas, S.; Maroso, F.; Karaiskou, N.; Carr, A.; Nielsen, E.E.; Bargelloni, L.; Triantafyllidis, A. Investigating genetic diversity and genomic signatures of hatchery-induced evolution in Gilthead seabream (Sparus aurata) populations. Diversity 2021, 13, 563. [Google Scholar] [CrossRef]
- Schlötterer, C.; Tobler, R.; Kofler, R.; Nolte, V. Sequencing pools of individuals-mining genome-wide polymorphism data without big funding. Nat. Rev. Genet. 2014, 15, 749–763. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bansal, V. A Statistical Method for the Detection of Variants from Next-Generation Resequencing of DNA Pools. Bioinformatics 2010, 26, i318–i324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karlsson, E.K.; Baranowska, I.; Wade, C.M.; Salmon Hillbertz, N.H.C.; Zody, M.C.; Anderson, N.; Biagi, T.M.; Patterson, N.; Pielberg, G.R.; Kulbokas, E.J.; et al. Efficient mapping of mendelian traits in dogs through genome-wide association. Nat. Genet. 2007, 39, 1321–1328. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A Flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [Green Version]
- Thomas, P.D.; Campbell, M.J.; Kejariwal, A.; Mi, H.; Karlak, B.; Daverman, R.; Diemer, K.; Muruganujan, A.; Narechania, A. PANTHER: A library of protein families and subfamilies indexed by function. Genome Res. 2003, 13, 2129–2141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Supek, F.; Bošnjak, M.; Škunca, N.; Šmuc, T. Revigo summarizes and visualizes long lists of Gene Ontology terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wade, C.M. Inbreeding and genetic diversity in dogs: Results from DNA analysis. Vet. J. 2011, 189, 183–188. [Google Scholar] [CrossRef]
- Arkush, K.D.; Giese, A.R.; Mendonca, H.L.; McBride, A.M.; Marty, G.D.; Hedrick, P.W. Resistance to three pathogens in the endangered winter-run Chinook salmon (Oncorhynchus tshawytscha): Effects of inbreeding and major histocompatibility complex genotypes. Can. J. Fish. Aquat. Sci. 2002, 59, 966–975. [Google Scholar] [CrossRef]
- Pante, M.J.R.; Gjerde, B.; McMillan, I. Effect of inbreeding on body weight at harvest in Rainbow trout, Oncorhynchus mykiss. Aquaculture 2001, 192, 201–211. [Google Scholar] [CrossRef]
- Rye, M.; Mao, I.L. Nonadditive genetic effects and inbreeding depression for body weight in Atlantic salmon (Salmo salar). Livest. Prod. Sci. 1998, 57, 15–22. [Google Scholar] [CrossRef]
- Gjerde, B.; Gunnes, K.; Gjedrem, T. Effect of inbreeding on survival and growth in Rainbow trout. Aquaculture 1983, 34, 327–332. [Google Scholar] [CrossRef]
- Perez-Enriquez, R.; Takagi, M.; Taniguchi, N. Genetic variability and pedigree tracing of a hatchery-reared stock of Red sea bream (Pagrus major) used for stock enhancement, based on microsatellite DNA markers. Aquaculture 1999, 173, 413–423. [Google Scholar] [CrossRef]
- Sundarrajan, L.; Unniappan, S. Small interfering RNA mediated knockdown of irisin suppresses food intake and modulates appetite regulatory peptides in Zebrafish. Gen. Comp. Endocrinol. 2017, 252, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhi, S.; Yang, G.; Qin, C.; Zhao, W.; Niu, M.; Zhang, W.; Tang, W.; Yan, X.; Zhang, Y.; et al. Molecular identification of fndc5 and effect of irisin on the glucose metabolism in Common carp (Cyprinus carpio). Gen. Comp. Endocrinol. 2021, 301, 113647. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Zhou, Y.; Yu, X.; Wang, J.; Luo, W.; Pang, M.; Tong, J. Genome-Wide Association study reveals SNPs and candidate genes related to growth and body shape in Bighead carp (Hypophthalmichthys nobilis). Mar. Biotechnol. 2022, 24, 1138–1147. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Fu, B.; Yu, X.; Chen, G.; Wang, J.; Luo, W.; Feng, Y.; Tong, J. Genome-Wide Association study reveals genomic regions and candidate genes for head size and shape in Bighead carp (Hypophthalmichthys nobilis). Aquaculture 2021, 539, 736648. [Google Scholar] [CrossRef]
- Bateman, A.; Bennett, H.P.J. The granulin gene family: From cancer to dementia. BioEssays 2009, 31, 1245–1254. [Google Scholar] [CrossRef] [PubMed]
- Ruan, D.; Zhuang, Z.; Ding, R.; Qiu, Y.; Zhou, S.; Wu, J.; Xu, C.; Hong, L.; Huang, S.; Zheng, E.; et al. Weighted single-step GWAS identified candidate genes associated with growth traits in a Duroc pig population. Genes 2021, 12, 117. [Google Scholar] [CrossRef] [PubMed]
- Mou, C.Y.; Li, Y.; Zhou, J.; Li, Q.; Zhou, B.; Wei, Z.; Luo, H.; Ke, H.Y.; Duan, Y.L.; Zhai, W.T.; et al. Genome-Wide Association study reveals growth-related markers and candidate genes for selection in Chinese longsnout catfish (Leiocassis longirostris). Aquaculture 2022, 560, 738513. [Google Scholar] [CrossRef]
- Carter, N.J.; Roach, Z.A.; Byrnes, M.M.; Zhu, Y. Adamts9 is necessary for ovarian development in Zebrafish. Gen. Comp. Endocrinol. 2019, 277, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, G.M.; Yáñez, J.M. Multi-Trait GWAS Using Imputed High-Density Genotypes from Whole-Genome Sequencing identifies genes associated with body traits in Nile tilapia. BMC Genom. 2021, 22, 57. [Google Scholar] [CrossRef] [PubMed]
- Prchal, M.; D’Ambrosio, J.; Lagarde, H.; Lallias, D.; Patrice, P.; François, Y.; Poncet, C.; Desgranges, A.; Haffray, P.; Dupont-Nivet, M.; et al. Genome-Wide Association study and genomic prediction of tolerance to acute hypoxia in Rainbow trout. Aquaculture 2023, 565, 739068. [Google Scholar] [CrossRef]
- Wang, L.Y.; Tian, Y.; Wen, H.S.; Yu, P.; Liu, Y.; Qi, X.; Gao, Z.C.; Zhang, K.Q.; Li, Y. SLC4 Gene family in Spotted sea bass (Lateolabrax maculatus): Structure, evolution, and expression profiling in response to alkalinity stress and salinity changes. Genes 2020, 11, 1271. [Google Scholar] [CrossRef]
- Sun, X.; Zhou, Y.; Zhang, R.; Wang, Z.; Xu, M.; Zhang, D.; Huang, J.; Luo, F.; Li, F.; Ni, Z.; et al. Dstyk mutation leads to congenital scoliosis-like vertebral malformations in Zebrafish via dysregulated MTORC1/TFEB pathway. Nat. Commun. 2020, 11, 479. [Google Scholar] [CrossRef] [Green Version]
- Ali, A.; Al-Tobasei, R.; Lourenco, D.; Leeds, T.; Kenney, B.; Salem, M. Genome-Wide identification of loci associated with growth in Rainbow trout. BMC Genom. 2020, 21, 209. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nuclei | No. of Fish | No. of Reads 1 | Depth (×) |
|---|---|---|---|
| Italy A1 | 30 | 320,381,051 | 57.69 |
| Italy A2 | 20 | 347,592,486 | 62.59 |
| Italy A3 | 30 | 330,168,763 | 59.45 |
| Italy B | 30 | 341,904,089 | 61.57 |
| Italy C | 30 | 339,891,846 | 61.21 |
| Nuclei/Populations | A1 | A2 | A3 | B | C | Farmed | Wild |
|---|---|---|---|---|---|---|---|
| A1 | 0 | ||||||
| A2 | 0.053 | 0 | |||||
| A3 | 0.052 | 0.032 | 0 | ||||
| B | 0.203 | 0.163 | 0.172 | 0 | |||
| C | 0.191 | 0.162 | 0.169 | 0.405 | 0 | ||
| Farmed | 0.05 | 0.04 | 0.04 | 0.163 | 0.166 | 0 | |
| Wild | 0.06 | 0.05 | 0.05 | 0.166 | 0.165 | 0.01 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bertolini, F.; Ribani, A.; Capoccioni, F.; Buttazzoni, L.; Bovo, S.; Schiavo, G.; Caggiano, M.; Rothschild, M.F.; Fontanesi, L. Whole Genome Sequencing Provides Information on the Genomic Architecture and Diversity of Cultivated Gilthead Seabream (Sparus aurata) Broodstock Nuclei. Genes 2023, 14, 839. https://doi.org/10.3390/genes14040839
Bertolini F, Ribani A, Capoccioni F, Buttazzoni L, Bovo S, Schiavo G, Caggiano M, Rothschild MF, Fontanesi L. Whole Genome Sequencing Provides Information on the Genomic Architecture and Diversity of Cultivated Gilthead Seabream (Sparus aurata) Broodstock Nuclei. Genes. 2023; 14(4):839. https://doi.org/10.3390/genes14040839
Chicago/Turabian StyleBertolini, Francesca, Anisa Ribani, Fabrizio Capoccioni, Luca Buttazzoni, Samuele Bovo, Giuseppina Schiavo, Massimo Caggiano, Max F. Rothschild, and Luca Fontanesi. 2023. "Whole Genome Sequencing Provides Information on the Genomic Architecture and Diversity of Cultivated Gilthead Seabream (Sparus aurata) Broodstock Nuclei" Genes 14, no. 4: 839. https://doi.org/10.3390/genes14040839