
Genomic Signatures of Local Adaptation under High Gene Flow in Lumpfish—Implications for Broodstock Provenance Sourcing and Larval Production

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Species
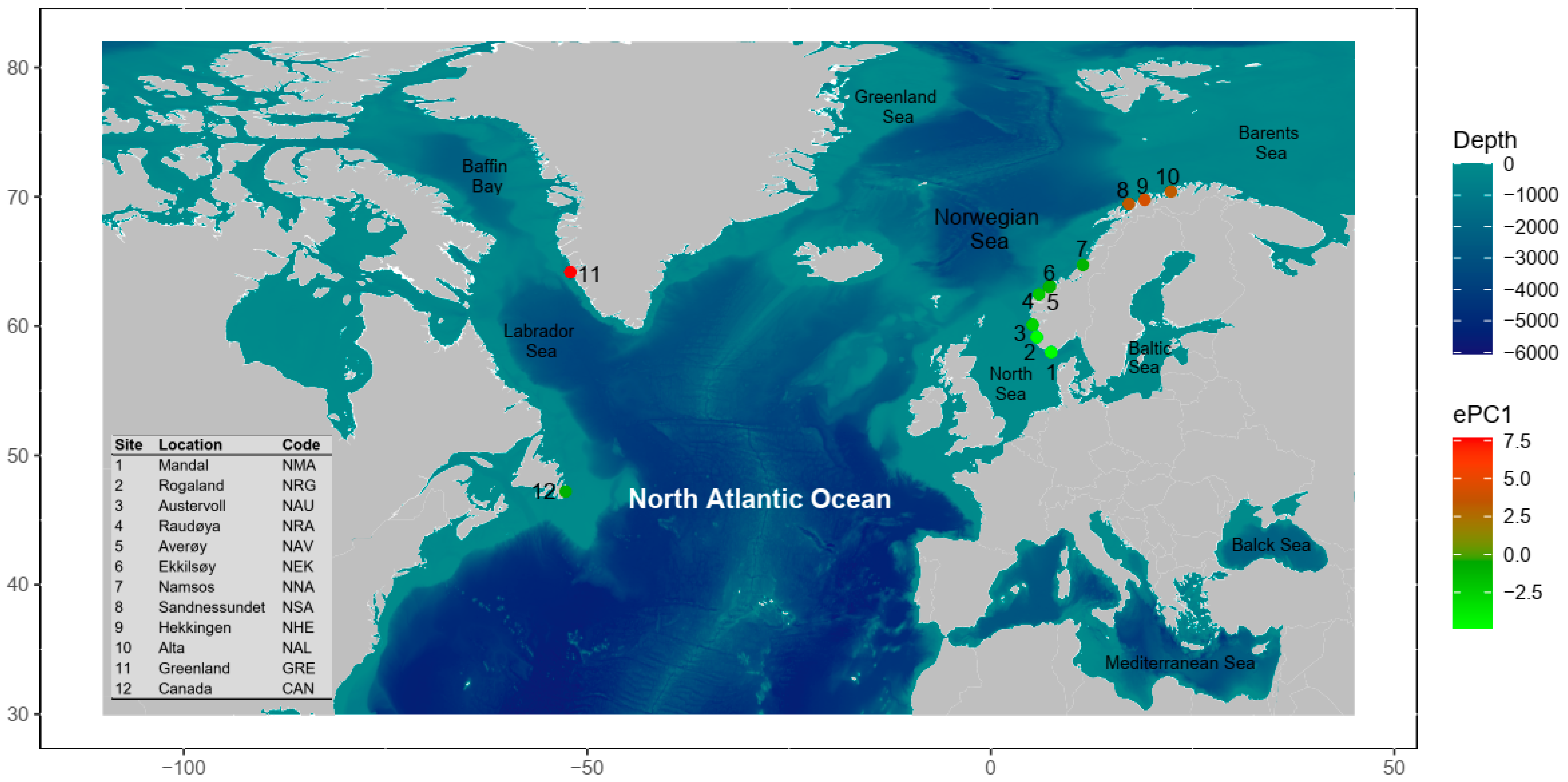
2.2. Sampling Regime
2.3. Genetic Data
2.3.1. DNA Extraction, 3RAD Library Preparation, and Sequencing


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Location | Code | Region | Latitude | Longitude | N | AP | AR | HO | HS | FIS (95% CI) | βWT (95% CI) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Mandal | NMA | Southern Norway | 57.99 | 7.48 | 10 (9) | 1 | 1.11 | 0.159 | 0.161 | 0.012 (0.005–0.020) | 0.023 (0.014–0.031) |
| Rogaland | NRG | Southern Norway | 59.15 | 5.70 | 11 (8) | 1 | 1.15 | 0.155 | 0.151 | −0.023 (−0.032–−0.015) | 0.102 (0.092–0.113) |
| Hardangerfjord | NHA | Southern Norway | 59.75 | 5.55 | 11 (0) | - | - | - | - | - | - |
| Austervoll | NAU | Southern Norway | 60.10 | 5.19 | 7 (7) | 0 | 1.17 | 0.158 | 0.165 | 0.041 (−0.011–0.013) | 0.022 (0.010–0.033) |
| Raudøya | NRA | Southern Norway | 62.45 | 5.97 | 5 (4) | 0 | 1.17 | 0.163 | 0.165 | 0.014 (0.003–0.025) | 0.020 (0.007–0.032) |
| Averøy | NAV | Southern Norway | 63.01 | 7.23 | 10 (10) | 4 | 1.16 | 0.162 | 0.163 | 0.006 (−0.001–0.014) | 0.028 (0.020–0.037) |
| Ekkilsøy | NEK | Southern Norway | 63.07 | 7.33 | 5 (4) | 0 | 1.16 | 0.160 | 0.156 | −0.022 (−0.034–−0.010) | 0.066 (0.054–0.079) |
| Namsos | NNA | Central Norway | 64.72 | 11.41 | 10 (10) | 1 | 1.17 | 0.162 | 0.165 | 0.017 (0.010–0.024) | 0.021 (0.013–0.030) |
| Sandnessundet | NSA | Northern Norway | 69.76 | 19.05 | 12 (11) | 3 | 1.16 | 0.159 | 0.156 | −0.020 (−0.027–−0.013) | 0.063 (0.055–0.071) |
| Hekkingen | NHE | Northern Norway | 69.45 | 17.10 | 11 (11) | 8 | 1.16 | 0.161 | 0.160 | −0.008 (−0.015–−0.001) | 0.047 (0.040–0.055) |
| Alta | NAL | Northern Norway | 70.40 | 22.31 | 15 (14) | 25 | 1.16 | 0.155 | 0.164 | 0.056 (0.050–0.063) | 0.026 (0.018–0.033) |
| Sermersooq | GRE | Western Greenland | 64.17 | −52.09 | 15 (15) | 243 | 1.15 | 0.151 | 0.154 | 0.020 (0.014–0.027) | 0.087 (0.074–0.099) |
| Canada | CAN | Eastern (Atlantic) Canada | 47.21 | −52.69 | 15 (14) | 180 | 1.13 | 0.135 | 0.135 | −0.001 (−0.008–0.006) | 0.188 (0.173–0.201) |
| Overall | 137 (117) | 38.8 | 1.15 | 0.157 | 0.159 | 0.008 (−0.004–0.013) | 0.058 (0.054–0.061) | ||||
2.3.2. SNP Discovery and Genotyping
2.3.3. Data Filtering
2.3.4. Genome Scans and Signatures of Selection
2.3.5. Genetic Diversity and Differentiation
2.3.6. Genetic Clustering and Connectivity
2.4. Environmental Data
2.5. Spatial Data
2.6. Seascape Genomics
2.7. Functional Annotation Analysis
3. Results
3.1. Genomic Datasets and Signatures of Selection
3.2. Genetic Diversity and Differentiation
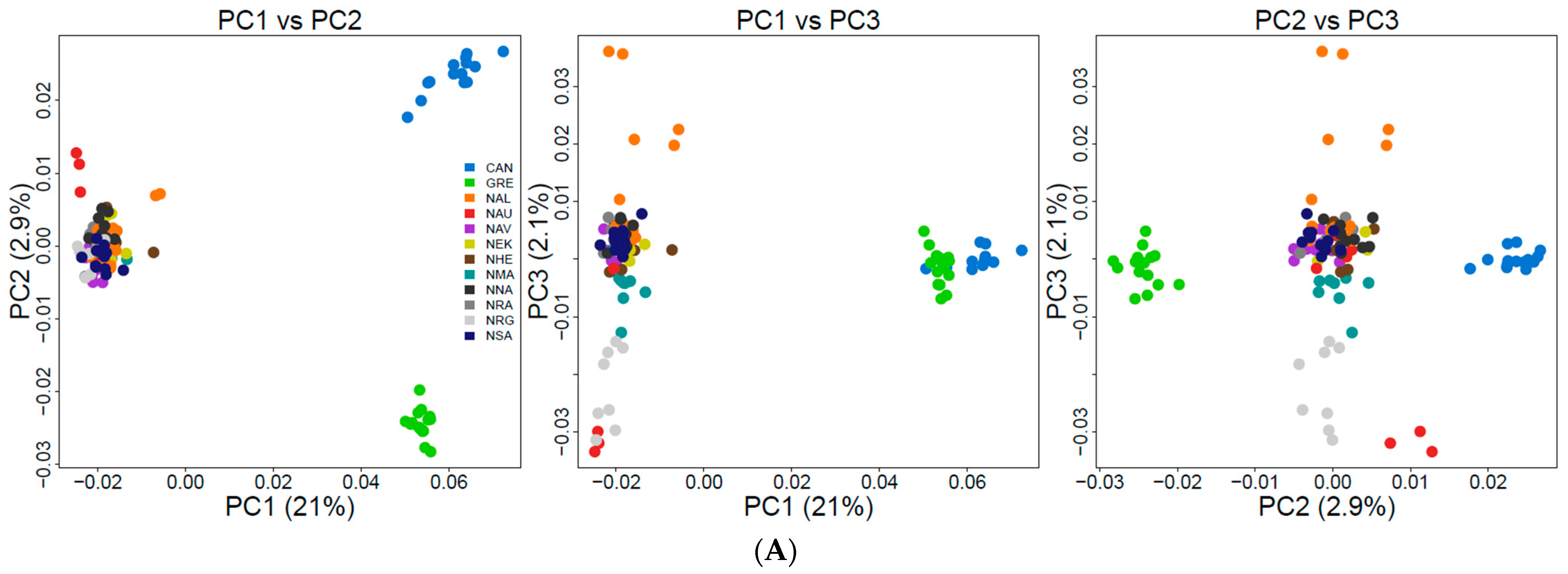
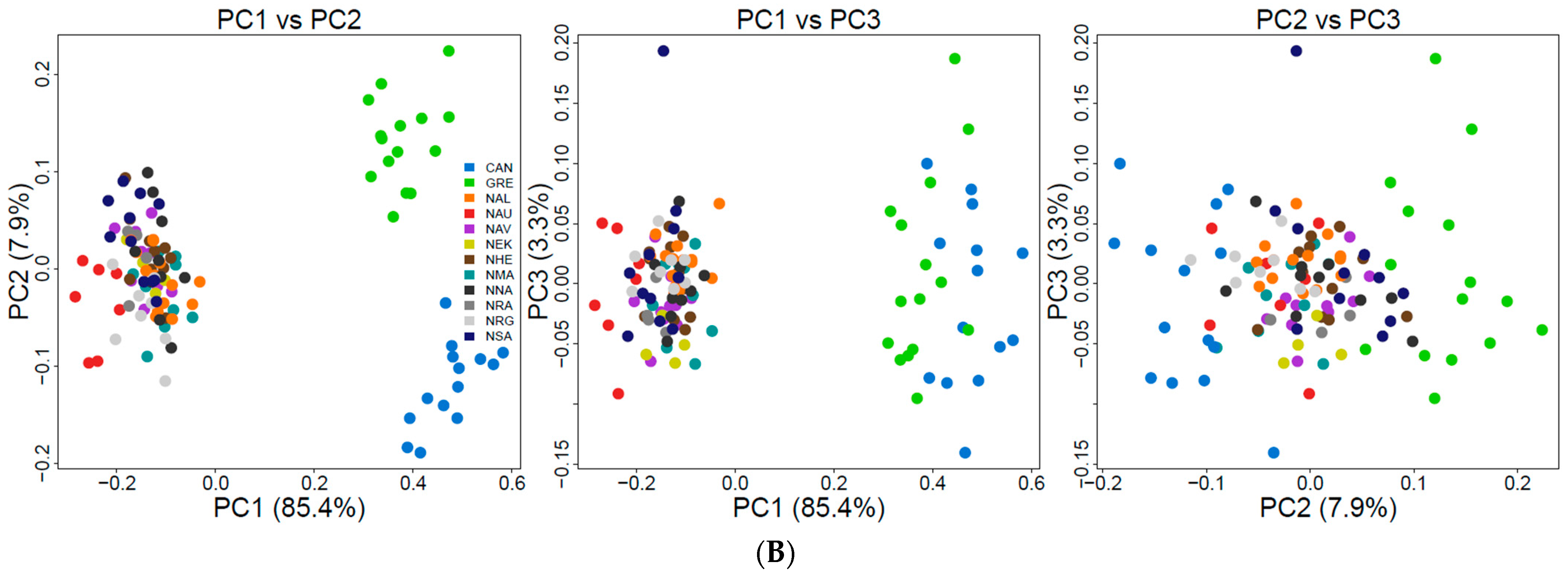
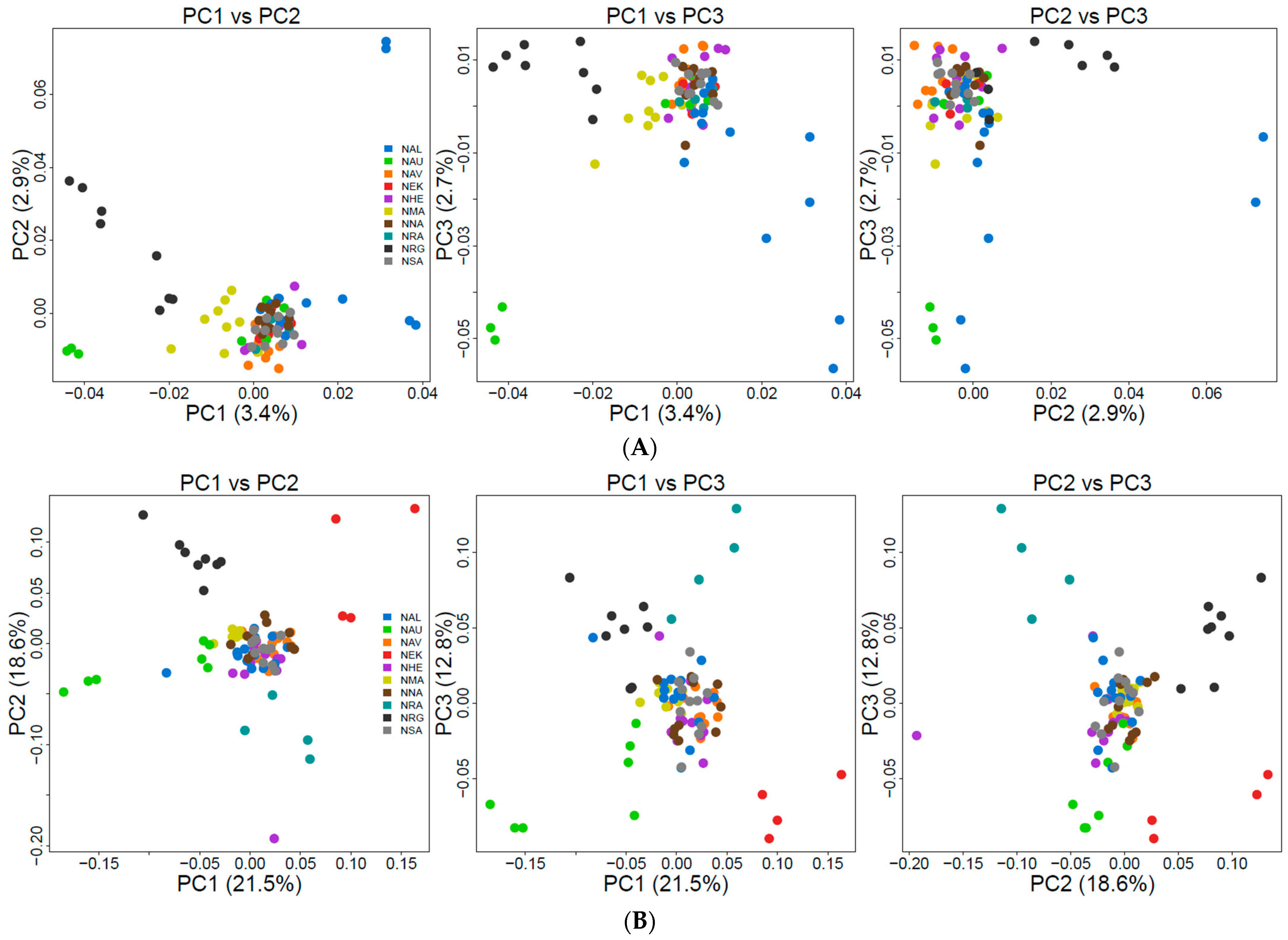
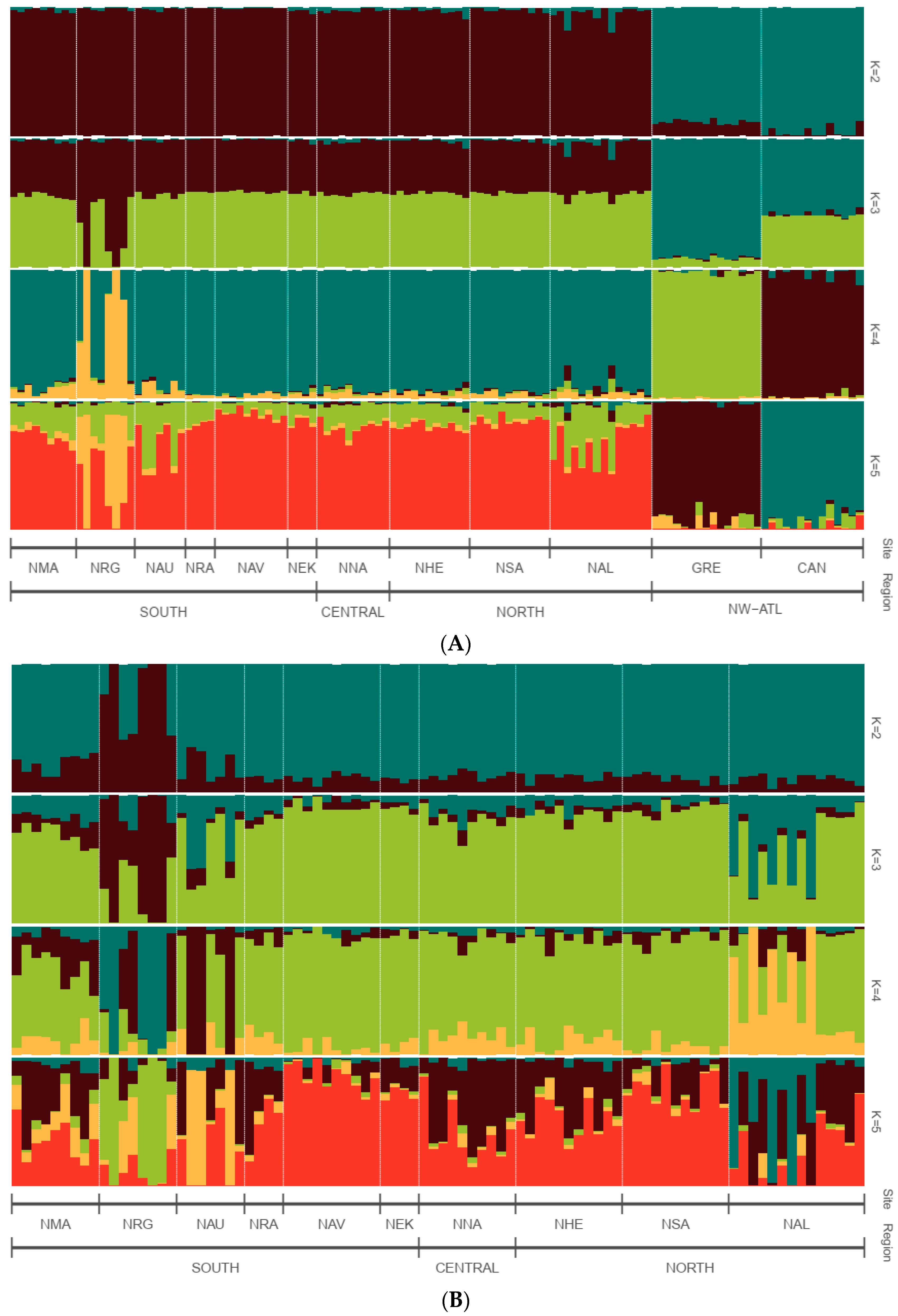
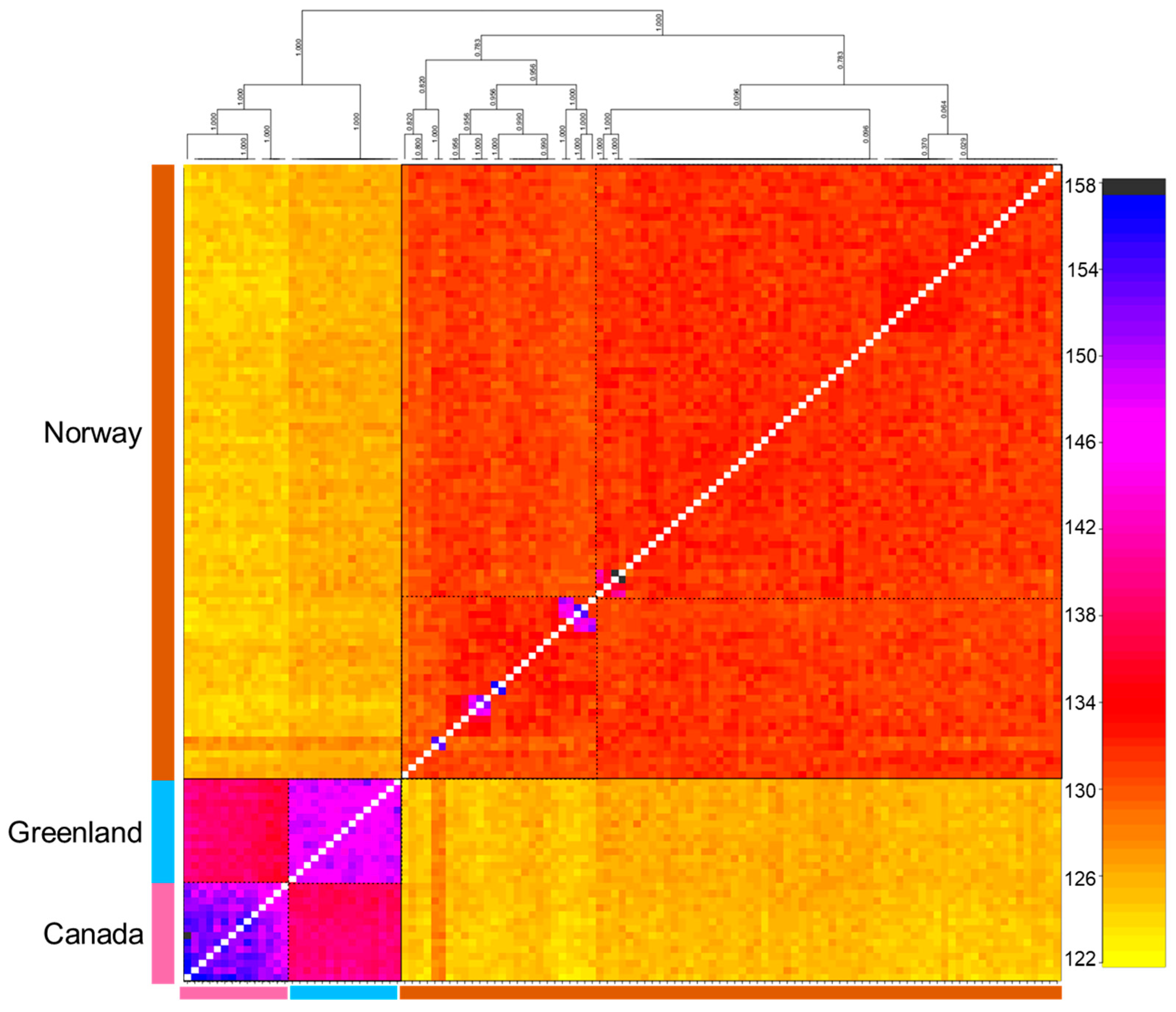
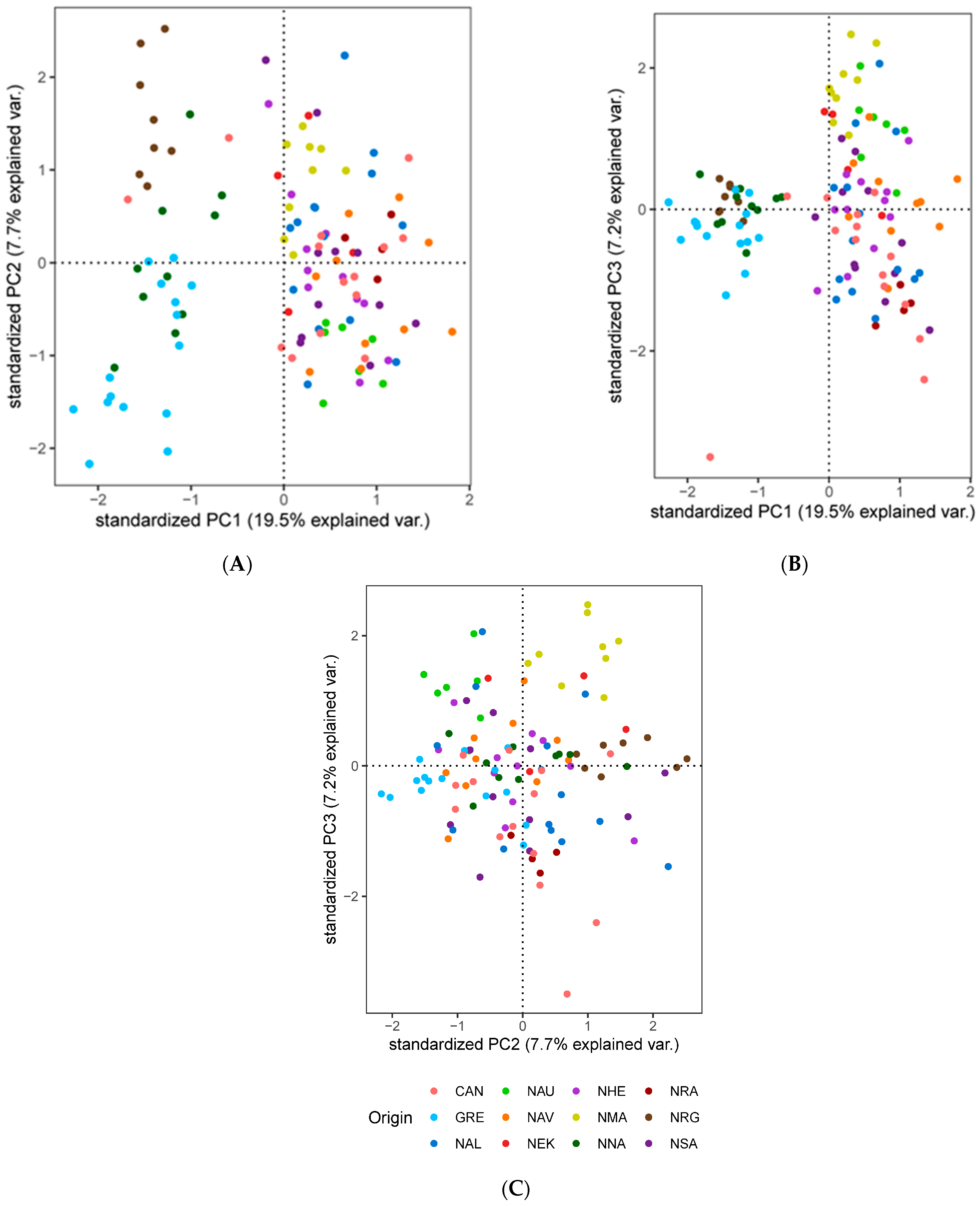
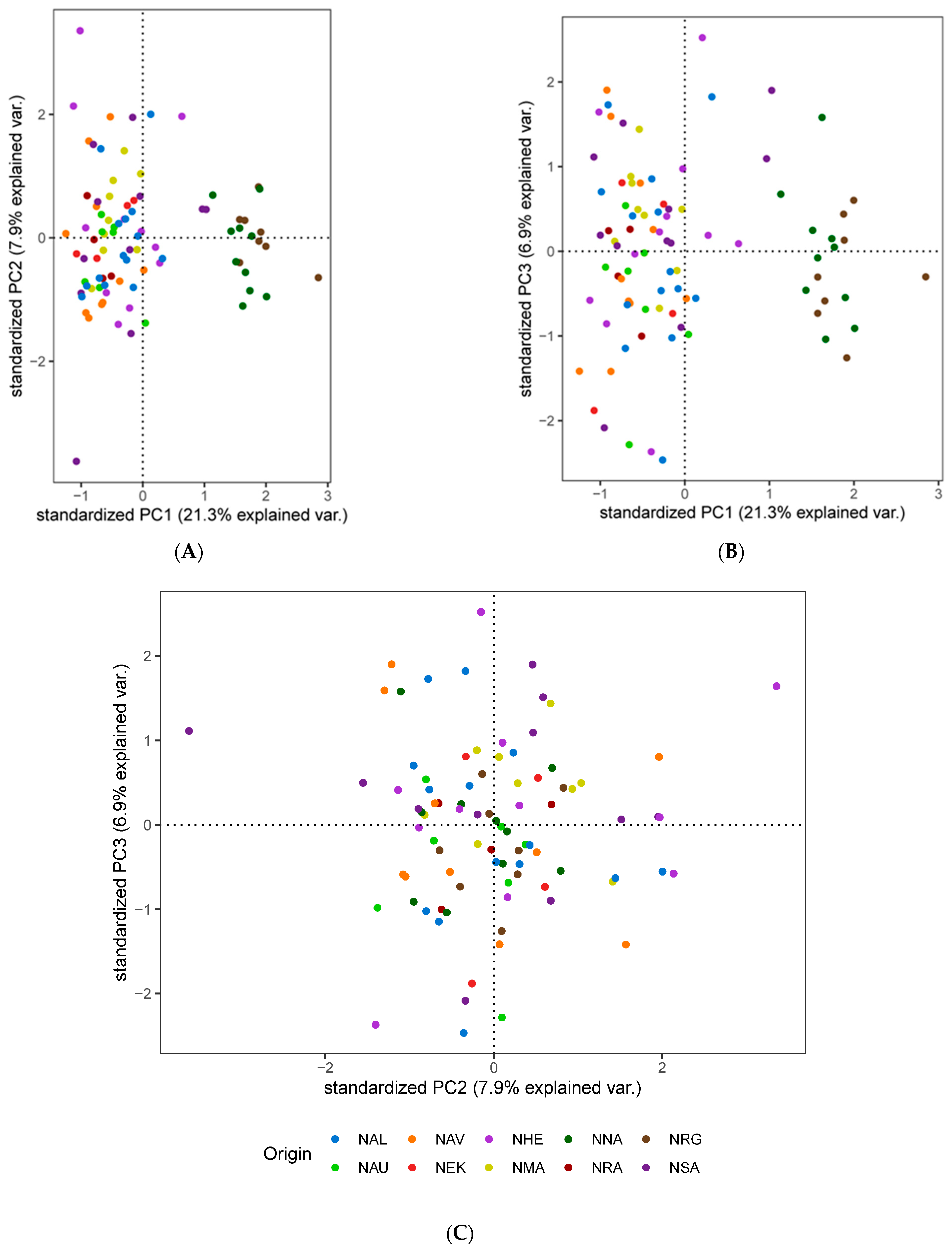
3.3. Population Genetic Structure and Connectivity
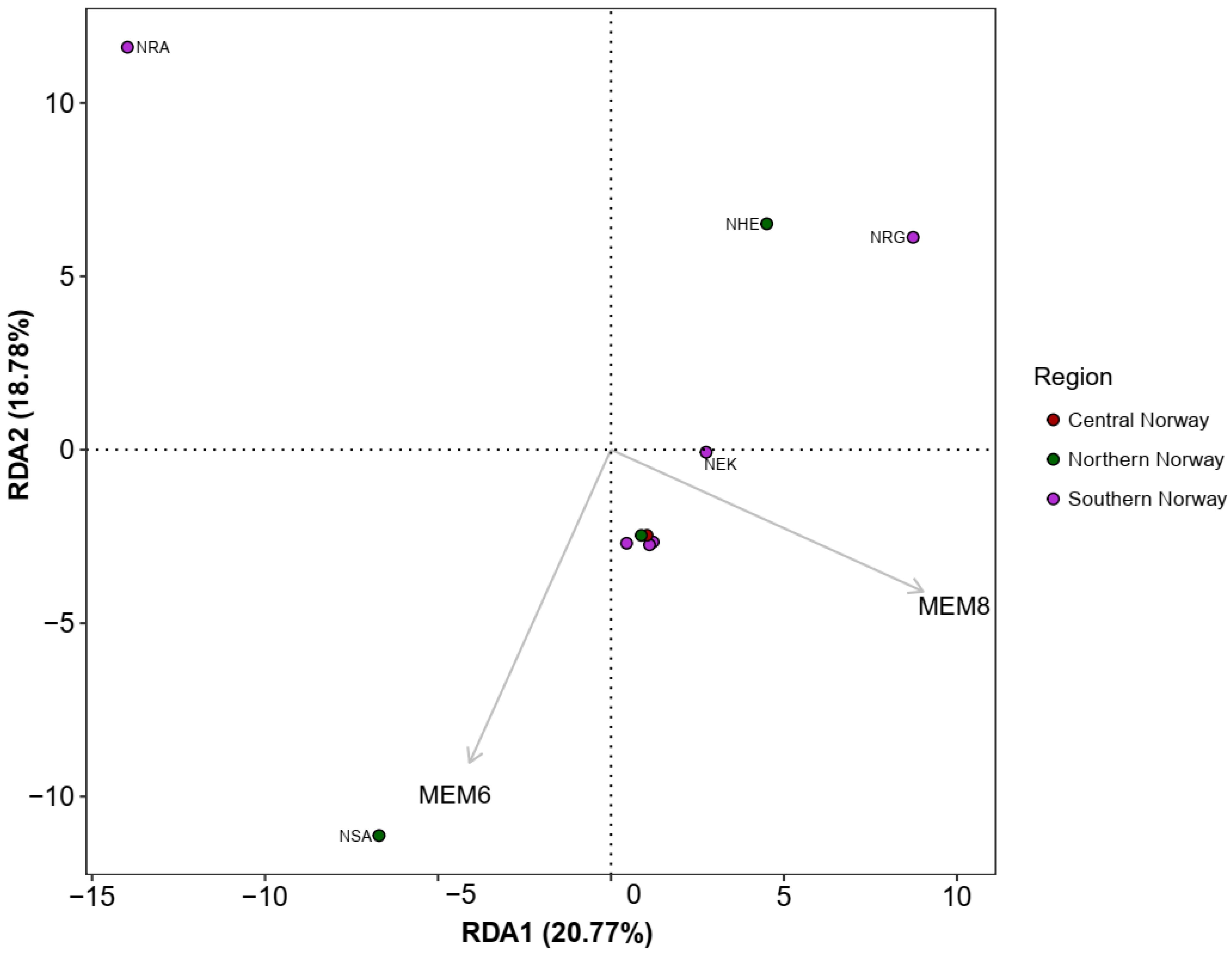
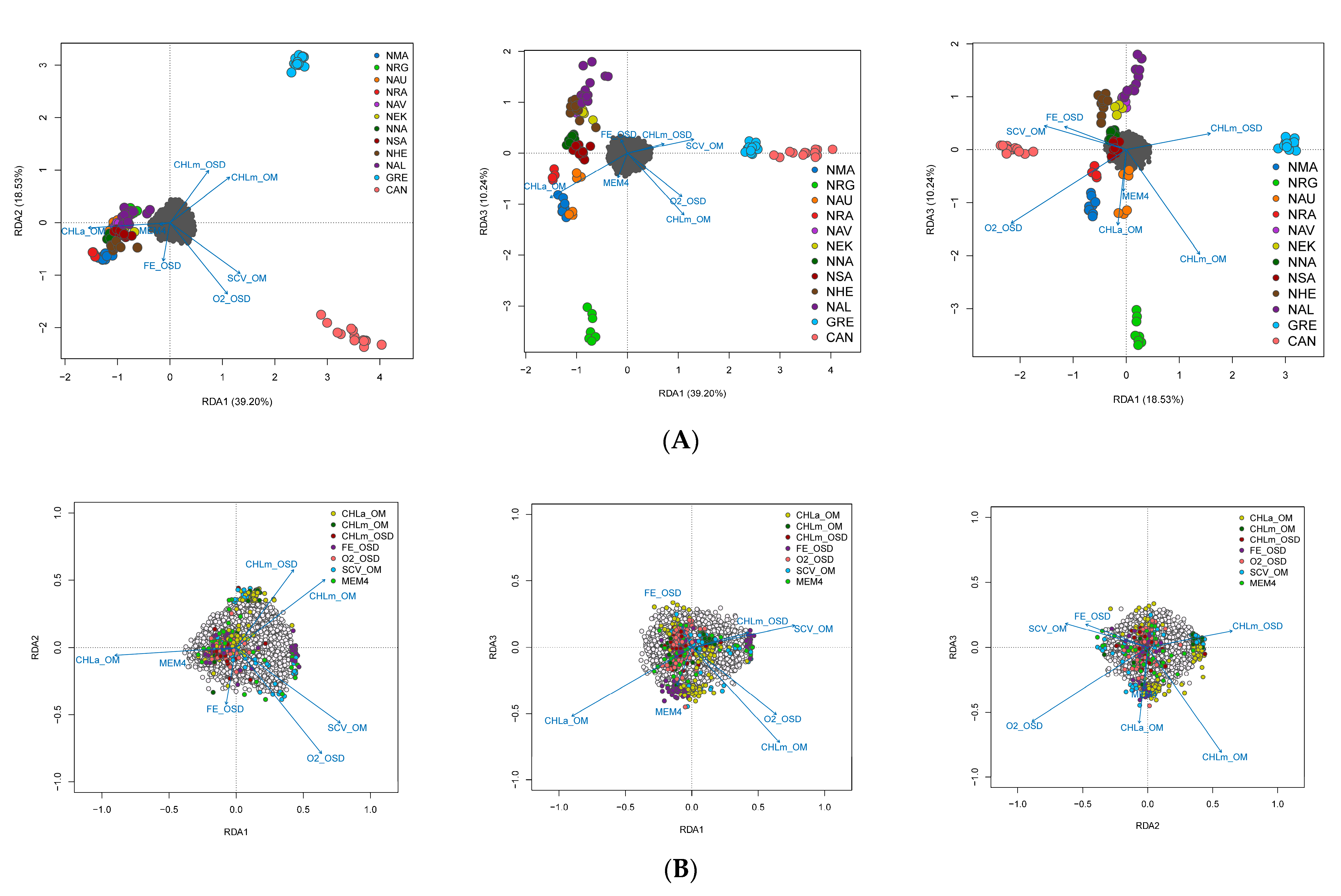
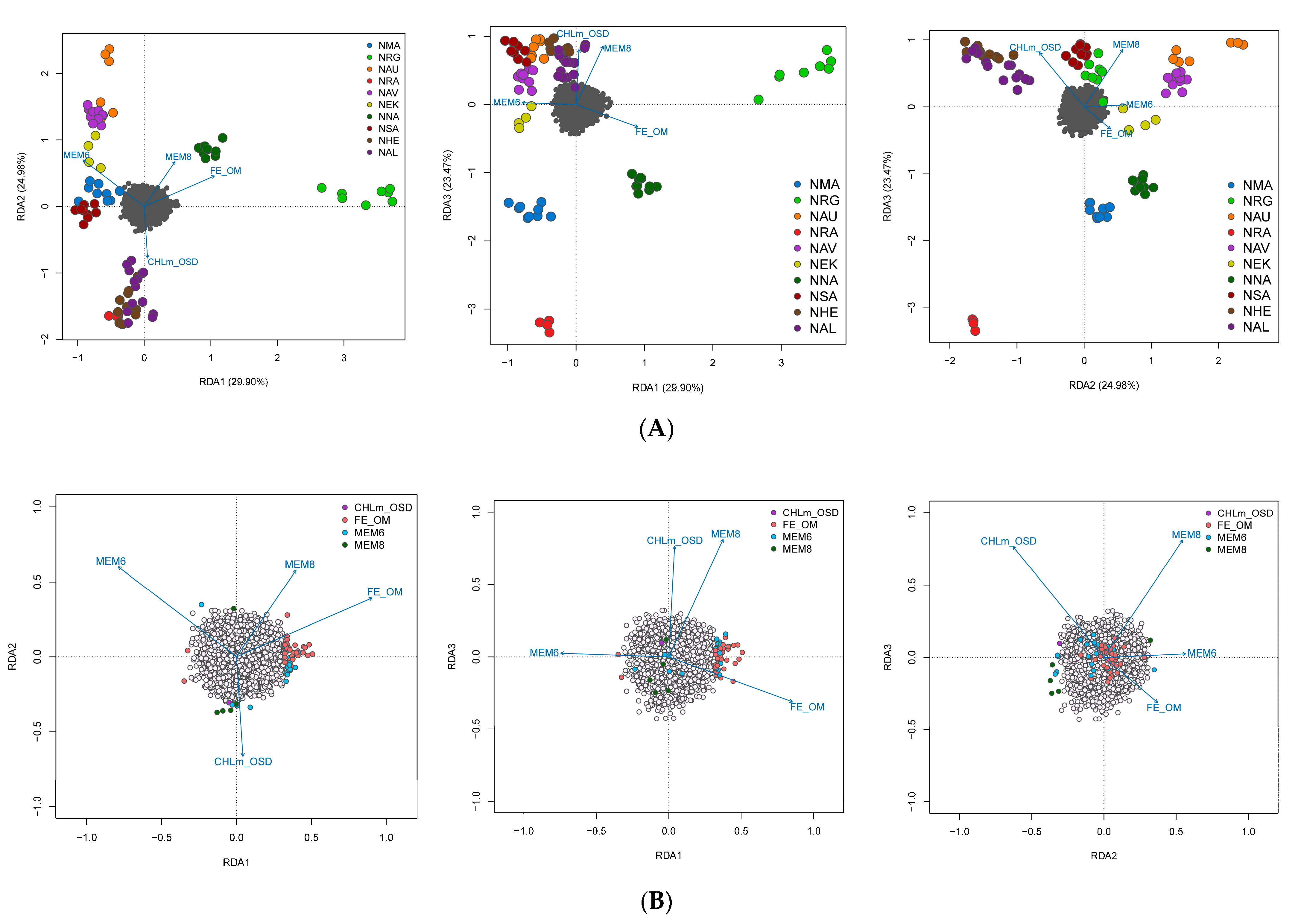
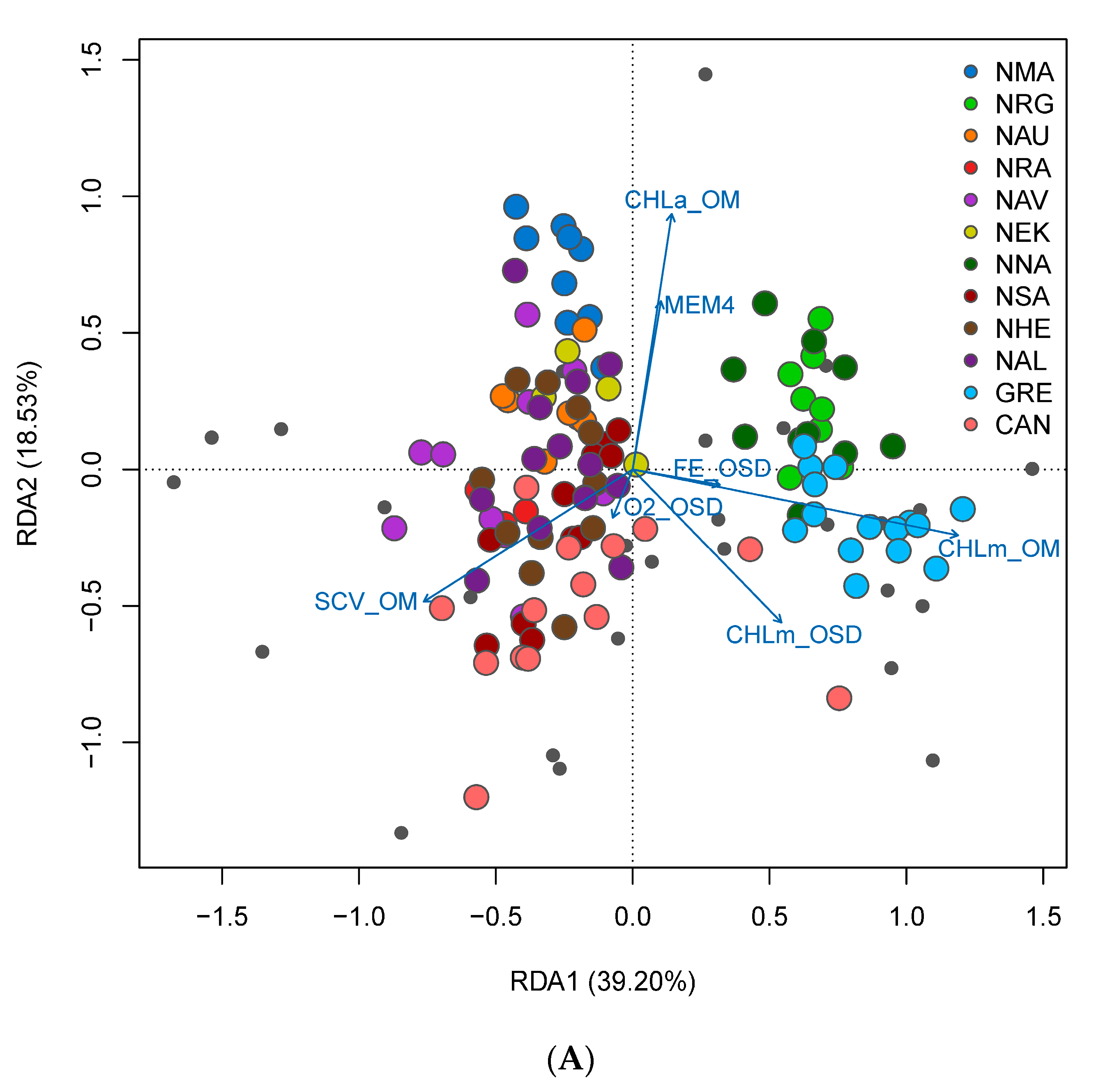
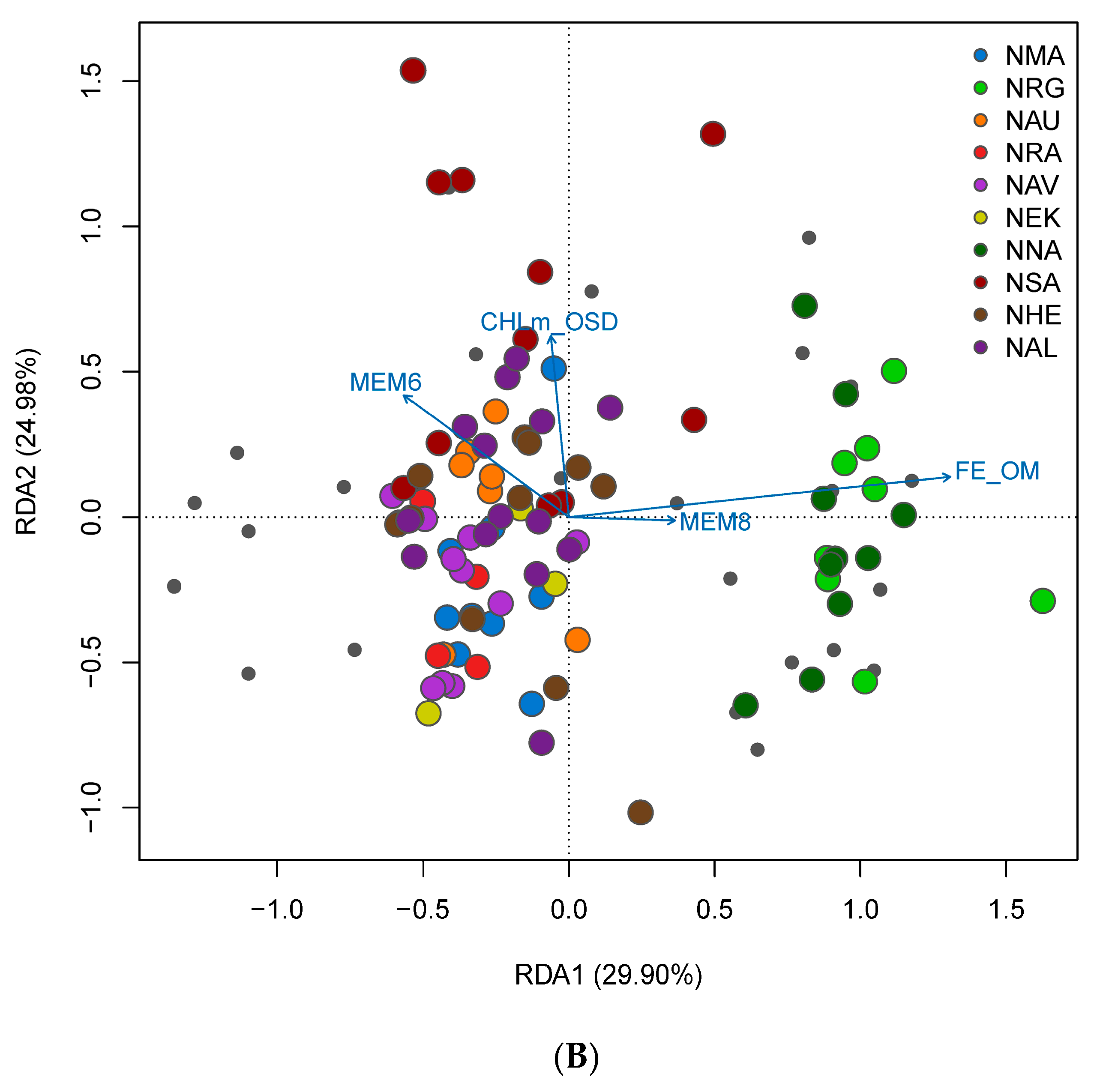
3.4. Spatial Structure and Environmental Associations
3.5. Functional Annotation
4. Discussion
4.1. Genome Scans for Outliers
4.2. Understanding the Population Structure of Lumpfish
4.3. Local Adaptation under High Gene Flow in Lumpfish
4.4. The Genomic Profile of Environmentally-Associated RAD Loci
4.5. Conclusions, Conservation, and Aquaculture Implications
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- FAO. The State of World Fisheries and Aquaculture 2022—Towards Blue Transformation; FAO: Rome, Italy, 2022. [Google Scholar] [CrossRef]
- Pradeepkiran, J.A. Aquaculture role in global food security with nutritional value: A review. Transl. Anim. Sci. 2019, 3, 903–910. [Google Scholar] [CrossRef] [PubMed]
- Misund, B.; Nygård, R. Big fish: Valuation of the world’s largest salmon farming companies. Mar. Resour. Econ. 2018, 33, 245–261. [Google Scholar] [CrossRef]
- Bailey, J.L.; Eggereide, S.S. Indicating sustainable salmon farming: The case of the new Norwegian aquaculture management scheme. Mar. Policy 2020, 117, 103925. [Google Scholar] [CrossRef]
- Iversen, A.; Asche, F.; Hermansen, Ø.; Nystøyl, R. Production cost and competitiveness in major salmon farming countries 2003–2018. Aquaculture 2020, 522, 735089. [Google Scholar] [CrossRef]
- Salmon Farming Industry Handbook. 2023. Available online: https://mowi.com/wp-content/uploads/2023/06/2023-Salmon-Farming-Industry-Handbook-2023.pdf (accessed on 5 August 2023).
- Cerbule, K.; Godfroid, J. Salmon louse (Lepeophtheirus salmonis (Krøyer)) control methods and efficacy in Atlantic salmon (Salmo salar (Linnaeus)) aquaculture: A literature review. Fishes 2020, 5, 11. [Google Scholar] [CrossRef]
- Imsland, A.K.D.; Reynolds, P. In lumpfish We Trust? The Efficacy of Lumpfish Cyclopterus lumpus to Control Lepeophtheirus salmonis Infestations on Farmed Atlantic Salmon: A Review. Fishes 2022, 7, 220. [Google Scholar] [CrossRef]
- Torrissen, O.; Jones, S.; Asche, F.; Guttormsen, A.; Skilbrei, O.T.; Nilsen, F.; Horsberg, T.E.; Jackson, D. Salmon lice—Impact on wild salmonids and salmon aquaculture. J. Fish Dis. 2013, 36, 171–194. [Google Scholar] [CrossRef]
- Bowers, J.M.; Mustafa, A.; Speare, D.J.; Conboy, G.A.; Brimacombe, M.; Sims, D.E.; Burka, J.F. The physiological response of Atlantic salmon, Salmo salar L., to a single experimental challenge with sea lice, Lepeophtheirus salmonis. J. Fish Dis. 2000, 23, 165–172. [Google Scholar] [CrossRef]
- Grave, K.; Horsberg, T.E.; Lunestad, B.T.; Litleskare, I. Consumption of drugs for sea lice infestations in Norwegian fish farms: Methods for assessment of treatment patterns and treatment rate. Dis. Aquat. Org. 2004, 60, 123–131. [Google Scholar] [CrossRef]
- Hamre, L.A.; Eichner, C.; Caipang, C.M.A.; Dalvin, S.T.; Bron, J.E.; Nilsen, F.; Boxshall, G.; Skern-Mauritzen, R. The Salmon Louse Lepeophtheirus salmonis (Copepoda: Caligidae) life cycle has only two Chalimus stages. PLoS ONE 2013, 8, e73539. [Google Scholar] [CrossRef]
- Contreras, M.; Karlsen, M.; Villar, M.; Olsen, R.H.; Leknes, L.M.; Furevik, A.; Yttredal, K.L.; Tartor, H.; Grove, S.; Alberdi, P.; et al. Vaccination with ectoparasite proteins involved in midgut function and blood digestion reduces salmon louse infestations. Vaccines 2020, 8, 32. [Google Scholar] [CrossRef] [PubMed]
- Bui, S.; Stien, L.H.; Nilsson, J.; Trengereid, H.; Oppedal, F. Efficiency and welfare impact of long-term simultaneous in situ management strategies for salmon louse reduction in commercial sea cages. Aquaculture 2020, 520, 734934. [Google Scholar] [CrossRef]
- Barrett, L.T.; Oppedal, F.; Robinson, N.; Dempster, T. Prevention not cure: A review of methods to avoid sea lice infestations in salmon aquaculture. Rev. Aquac. 2020, 12, 2527–2543. [Google Scholar] [CrossRef]
- Johnson, S.C.; Constible, J.M.; Richard, J. Laboratory investigations on the efficacy of hydrogen peroxide against the salmon louse Lepeophtheirus salmonis and its toxicological and histopathological effects on Atlantic salmon Salmo salar and Chinook salmon Oncorhynchus tshawytscha. Dis. Aquat. Org. 1993, 17, 197–204. [Google Scholar] [CrossRef]
- Overton, K.; Dempster, T.; Oppedal, F.; Kristiansen, T.S.; Gismervik, K.; Stien, L.H. Salmon lice treatments and salmon mortality in Norwegian aquaculture: A review. Rev. Aquac. 2019, 11, 1398–1417. [Google Scholar] [CrossRef]
- Skiftesvik, A.B.; Bjelland, R.M.; Durif, C.M.; Johansen, I.S.; Browman, H.I. Delousing of Atlantic salmon (Salmo salar) by cultured vs. wild ballan wrasse (Labrus bergylta). Aquaculture 2013, 402, 113–118. [Google Scholar] [CrossRef]
- Imsland, A.K.; Reynolds, P.; Eliassen, G.; Hangstad, T.A.; Foss, A.; Vikingstad, E.; Elvegård, T.A. The use of lumpfish (Cyclopterus lumpus L.) to control sea lice (Lepeophtheirus salmonis Krøyer) infestations in intensively farmed Atlantic salmon (Salmo salar L.). Aquaculture 2014, 424, 18–23. [Google Scholar] [CrossRef]
- Overton, K.; Barrett, L.T.; Oppedal, F.; Kristiansen, T.S.; Dempster, T. Sea lice removal by cleaner fish in salmon aquaculture: A review of the evidence base. Aquac. Environ. Interact. 2020, 12, 31–44. [Google Scholar] [CrossRef]
- Bolton-Warberg, M. An overview of cleaner fish use in Ireland. J. Fish Dis. 2018, 41, 935–939. [Google Scholar] [CrossRef]
- Treasurer, J.; Prickett, R.; Zietz, M.; Hempleman, C.; Garcia de Leaniz, C. Cleaner fish rearing and deployment in the UK. In Cleaner Fish Biology and Aquaculture Applications; Treasurer, J.W., Ed.; 5M Publishing Ltd.: Sheffield, UK, 2018; pp. 376–391. [Google Scholar]
- Imsland, A.K.D.; Reynolds, P.; Hangstad, T.A.; Kapari, L.; Maduna, S.N.; Hagen, S.B.; Jónsdóttir, Ó.D.B.; Spetland, F.; Lindberg, K.S. Quantification of grazing efficacy, growth and health score of different lumpfish (Cyclopterus lumpus L.) families: Possible size and gender effects. Aquaculture 2021, 530, 735925. [Google Scholar] [CrossRef]
- Norway Directorate of Fisheries. Akvakulturstatistikk: Rensefisk. 2022. Available online: https://www.fiskeridir.no/Akvakultur/Tall-og-analyse/Akvakulturstatistikk-tidsserier/Rensefisk (accessed on 15 December 2022).
- Holmyard, N. Lumpfish Production Becoming Big Business in Norway. SeafoodSource. 2018. Available online: https://www.seafoodsource.com/news/aquaculture/lumpfish-production-becoming-big-business-in-norway (accessed on 5 August 2022).
- Sae-Lim, P.; Khaw, H.L.; Nielsen, H.M.; Puvanendran, V.; Hansen, Ø.; Mortensen, A. Genetic variance for uniformity of body weight in lumpfish (Cyclopterus lumpus) used a double hierarchical generalized linear model. Aquaculture 2020, 514, 734515. [Google Scholar] [CrossRef]
- Jónsdóttir, Ó.D.B.; Imsland, A.K.; Kennedy, J. Lumpfish biology and population genetics. In Cleaner Fish Biology and Aquaculture Applications; Treasurer, J., Ed.; 5M Publishing Ltd.: Sheffield, UK, 2018. [Google Scholar]
- Jónsdóttir, Ó.D.B.; Schregel, J.; Hagen, S.B.; Tobiassen, C.; Aarnes, S.G.; Imsland, A.K. Population genetic structure of lumpfish along the Norwegian coast: Aquaculture implications. Aquac. Int. 2018, 26, 49–60. [Google Scholar] [CrossRef]
- Pampoulie, C.; Skirnisdottir, S.; Olafsdottir, G.; Helyar, S.J.; Thorsteinsson, V.; Jónsson, S.; Fréchet, A.; Durif, C.M.F.; Sherman, S.; Lampart-Kałużniacka, M.; et al. Genetic structure of the lumpfish Cyclopterus lumpus across the North Atlantic. ICES J. Mar. Sci. 2014, 71, 2390–2397. [Google Scholar] [CrossRef]
- Garcia-Mayoral, E.; Olsen, M.; Hedeholm, R.; Post, S.; Nielsen, E.E.; Bekkevold, D. Genetic structure of West Greenland populations of lumpfish Cyclopterus lumpus. J. Fish Biol. 2016, 89, 2625–2642. [Google Scholar] [CrossRef]
- Whittaker, B.A.; Consuegra, S.; Garcia de Leaniz, C. Genetic and phenotypic differentiation of lumpfish (Cyclopterus lumpus) across the North Atlantic: Implications for conservation and aquaculture. PeerJ 2018, 6, e5974. [Google Scholar] [CrossRef]
- Jónsdóttir, Ó.D.B.; Gíslason, D.; Ólafsdóttir, G.; Maduna, S.N.; Hagen, S.B.; Reynolds, P.; Sveinsson, S.; Imsland, A.K. Lack of population genetic structure of lumpfish along the Norwegian coast: A reappraisal based on EST-STRs analyses. Aquaculture 2022, 555, 738230. [Google Scholar] [CrossRef]
- Jansson, E.; Faust, E.; Bekkevold, D.; Quintela, M.; Durif, C.; Halvorsen, K.T.; Dahle, G.; Pampoulie, C.; Kennedy, J.; Whittaker, B.; et al. Global, regional, and cryptic population structure in a high gene-flow transatlantic fish. PLoS ONE 2023, 18, e0283351. [Google Scholar] [CrossRef]
- Williams, G.C. Adaptation and Natural Selection; Princeton University Press: Princeton, NJ, USA, 1966. [Google Scholar]
- Kawecki, T.J.; Ebert, D. Conceptual issues in local adaptation. Ecol. Lett. 2004, 7, 1225–1241. [Google Scholar] [CrossRef]
- Blanquart, F.; Kaltz, O.; Nuismer, S.L.; Gandon, S. A practical guide to measuring local adaptation. Ecol. Lett. 2013, 16, 1195–1205. [Google Scholar] [CrossRef]
- Savolainen, O.; Lascoux, M.; Merilä, J. Ecological genomics of local adaptation. Nat. Rev. Genet. 2013, 14, 807–820. [Google Scholar] [CrossRef]
- Fraser, D.J.; Cook, A.M.; Eddington, J.D.; Bentzen, P.; Hutchings, J.A. Mixed evidence for reduced local adaptation in wild salmon resulting from interbreeding with escaped farmed salmon: Complexities in hybrid fitness. Evol. Appl. 2008, 1, 501–512. [Google Scholar] [CrossRef] [PubMed]
- Bourret, V.; O’Reilly, P.T.; Carr, J.W.; Berg, P.R.; Bernatchez, L. Temporal change in genetic integrity suggests loss of local adaptation in a wild Atlantic salmon (Salmo salar) population following introgression by farmed escapees. Heredity 2011, 106, 500–510. [Google Scholar] [CrossRef] [PubMed]
- Antoniou, A.; Manousaki, T.; Ramírez, F.; Cariani, A.; Cannas, R.; Kasapidis, P.; Magoulas, A.; Albo-Puigserver, M.; Lloret-Lloret, E.; Bellido, J.M.; et al. Sardines at a junction: Seascape genomics reveals ecological and oceanographic drivers of variation in the NW Mediterranean Sea. Mol. Ecol. 2023, 32, 1608–1628. [Google Scholar] [CrossRef] [PubMed]
- Levins, R. The theory of fitness in a heterogeneous environment. IV. The adaptive significance of gene flow. Evol. Int. J. Org. Evol. 1964, 18, 635–638. [Google Scholar] [CrossRef]
- Endler, J.A. Gene flow and population differentiation. Science 1973, 179, 243–250. [Google Scholar] [CrossRef]
- Garant, D.A.N.Y.; Forde, S.E.; Hendry, A.P. The multifarious effects of dispersal and gene flow on contemporary adaptation. Funct. Ecol. 2007, 21, 434–443. [Google Scholar] [CrossRef]
- Fitzpatrick, S.W.; Gerberich, J.C.; Kronenberger, J.A.; Angeloni, L.M.; Funk, W.C. Locally adapted traits maintained in the face of high gene flow. Ecol. Lett. 2015, 18, 37–47. [Google Scholar] [CrossRef]
- Tigano, A.; Friesen, V.L. Genomics of local adaptation with gene flow. Mol. Ecol. 2016, 25, 2144–2164. [Google Scholar] [CrossRef]
- Haldane, J.B.S. A mathematical theory of natural and artificial selection (Part VI, Isolation.). In Mathematical Proceedings of the Cambridge Philosophical Society; Cambridge University Press: Cambridge, UK, 1930; Volume 26, pp. 220–230. [Google Scholar]
- Lenormand, T. Gene flow and the limits to natural selection. Trends Ecol. Evol. 2002, 17, 183–189. [Google Scholar] [CrossRef]
- Allendorf, F.W.; Luikart, G. Conservation and the Genetics of Populations; Blackwell Publishing: Oxford, UK, 2007. [Google Scholar]
- Bridle, J.R.; Vines, T.H. Limits to evolution at range margins: When and why does adaptation fail? Trends Ecol. Evol. 2007, 22, 140–147. [Google Scholar] [CrossRef]
- Tallmon, D.A.; Luikart, G.; Waples, R.S. The alluring simplicity and complex reality of genetic rescue. Trends Ecol. Evol. 2004, 19, 489–496. [Google Scholar] [CrossRef]
- Bontrager, M.; Angert, A.L. Gene flow improves fitness at a range edge under climate change. Evol. Lett. 2018, 3, 55–68. [Google Scholar] [CrossRef]
- Clark, J.D.; Benham, P.M.; Maldonado, J.E.; Luther, D.A.; Lim, H.C. Maintenance of local adaptation despite gene flow in a coastal songbird. Evol. Int. J. Org. Evol. 2022, 76, 1481–1494. [Google Scholar] [CrossRef]
- Salloum, P.M.; Santure, A.W.; Lavery, S.D.; de Villemereuil, P. Finding the adaptive needles in a population-structured haystack: A case study in a New Zealand mollusc. J. Anim. Ecol. 2022, 91, 1209–1221. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, A.; Griffin, P.; Dillon, S.; Catullo, R.; Rane, R.; Byrne, M.; Jordan, R.; Oakeshott, J.; Weeks, A.; Joseph, L.; et al. A framework for incorporating evolutionary genomics into biodiversity conservation and management. Clim. Chang. Responses 2015, 2, 1. [Google Scholar] [CrossRef]
- Bayona-Vásquez, N.J.; Glenn, T.C.; Kieran, T.J.; Pierson, T.W.; Hoffberg, S.L.; Scott, P.A.; Bentley, K.E.; Finger, J.W.; Louha, S.; Troendle, N.; et al. Adapterama III: Quadruple-indexed, double/triple-enzyme RADseq libraries (2RAD/3RAD). PeerJ 2019, 7, e7724. [Google Scholar] [CrossRef] [PubMed]
- Davenport, J. Synopsis of Biological Data on the Lumpsucker, Cyclopterus lumpus (Linnaeus, 1758); Food & Agriculture Organization: Rome, Italy, 1985; Volume 147. [Google Scholar]
- Bañón, R.; Garazo, A.; Fernández, A. Short communication Note about the presence of the lumpsucker Cyclopterus lumpus (Teleostei, Cyclopteridae) in Galician waters (NW Spain). J. Appl. Ichthyol. 2007, 24, 108–109. [Google Scholar] [CrossRef]
- Voskoboinikova, O.S.; Kudryavtseva, O.Y.; Orlov, A.M.; Orlova, S.Y.; Nazarkin, M.V.; Chernova, N.V.; Maznikova, O.A. Relationships and evolution of lumpsuckers of the family Cyclopteridae (Cottoidei). J. Ichthyol. 2020, 60, 154–181. [Google Scholar] [CrossRef]
- Kennedy, J.; Durif, C.M.F.; Florin, A.-B.; Fréchet, A.; Gauthier, J.; Hüssy, K.; Jónsson, S.; Ólafsson, H.G.; Post, S.; Hedeholm, R.B. A brief history of lumpfishing, assessment, and management across the North Atlantic. ICES J. Mar. Sci. 2019, 76, 181–191. [Google Scholar] [CrossRef]
- Kennedy, J.; Jónsson, S.Þ.; Ólafsson, H.G.; Kasper, J.M. Observations of vertical movements and depth distribution of migrating female lumpfish (Cyclopterus lumpus) in Iceland from data storage tags and trawl surveys. ICES J. Mar. Sci. 2016, 73, 1160–1169. [Google Scholar] [CrossRef]
- Zvetkov, V.I.; Kalyakina, N.M. Relationships between White Sea fish and macrophytes. Ecology 1987, 6, 40–43. [Google Scholar]
- Kudryavzeva, O.J. The Lumpsucker (Cyclopterus lumpus) in the Barents Sea and Adjusted Waters. Ph.D. Thesis, Nauka Press, Moskow, Russia, 2008. [Google Scholar]
- Gade, H.G. (Ed.) Features of fjord and ocean interaction. In The Nordic Seas; Springer: New York, NY, USA, 1986; pp. 183–190. [Google Scholar]
- Sætre, R. (Ed.) The Norwegian Coastal Current: Oceanography and Climate; Fagbokforlaget: Oslo, Norway, 2007. [Google Scholar]
- Skagseth, Ø.; Drinkwater, K.F.; Terrile, E. Wind-and buoyancy-induced transport of the Norwegian Coastal Current in the Barents Sea. J. Geophys. Res. Ocean. 2011, 116, C08007. [Google Scholar] [CrossRef]
- Peterson, B.K.; Weber, J.N.; Kay, E.H.; Fisher, H.S.; Hoekstra, H.E. Double digest RADseq: An inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoS ONE 2012, 7, e37135. [Google Scholar] [CrossRef] [PubMed]
- Graham, C.F.; Glenn, T.C.; McArthur, A.G.; Boreham, D.R.; Kieran, T.; Lance, S.; Manzon, R.G.; Martino, J.A.; Pierson, T.; Rogers, S.M.; et al. Impacts of degraded DNA on restriction enzyme associated DNA sequencing (RADSeq). Mol. Ecol. Resour. 2015, 15, 1304–1315. [Google Scholar] [CrossRef]
- Hoffberg, S.L.; Kieran, T.J.; Catchen, J.M.; Devault, A.; Faircloth, B.C.; Mauricio, R.; Glenn, T.C. RADcap: Sequence capture of dual-digest RADseq libraries with identifiable duplicates and reduced missing data. Mol. Ecol. Resour. 2016, 16, 1264–1278. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 29 June 2023).
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef]
- Rochette, N.C.; Rivera-Colón, A.G.; Catchen, J.M. Stacks 2: Analytical methods for paired-end sequencing improve RADseq-based population genomics. Mol. Ecol. 2019, 28, 4737–4754. [Google Scholar] [CrossRef]
- Holborn, M.K.; Einfeldt, A.L.; Kess, T.; Duffy, S.J.; Messmer, A.M.; Langille, B.L.; Brachmann, M.K.; Gauthier, J.; Bentzen, P.; Knutsen, T.M.; et al. Reference genome of lumpfish Cyclopterus lumpus Linnaeus provides evidence of male heterogametic sex determination through the AMH pathway. Mol. Ecol. Resour. 2022, 22, 1427–1439. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Okonechnikov, K.; Conesa, A.; García-Alcalde, F. Qualimap 2: Advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics 2016, 32, 292–294. [Google Scholar] [CrossRef]
- McKinney, G.J.; Waples, R.K.; Seeb, L.W.; Seeb, J.E. Paralogs are revealed by proportion of heterozygotes and deviations in read ratios in genotyping-by-sequencing data from natural populations. Mol. Ecol. Resour. 2017, 17, 656–669. [Google Scholar] [CrossRef] [PubMed]
- Dorant, Y.; Cayuela, H.; Wellband, K.; Laporte, M.; Rougemont, Q.; Mérot, C.; Normandeau, E.; Rochette, R.; Bernatchez, L. Copy number variants outperform SNPs to reveal genotype-temperature association in a marine species. Mol. Ecol. 2020, 29, 4765–4782. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Benyamin, B.; McEvoy, B.P.; Gordon, S.; Henders, A.K.; Nyholt, D.R.; Madden, P.A.; Heath, A.C.; Martin, N.G.; Montgomery, G.W. Common SNPs explain a large proportion of the heritability for human height. Nat. Genet. 2010, 42, 565–569. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. 1000 Genomes Project Analysis Group. (Aug 1, 2011) The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Manichaikul, A.; Mychaleckyj, J.C.; Rich, S.S.; Daly, K.; Sale, M.; Chen, W.M. Robust relationship inference in genome-wide association studies. Bioinformatics 2010, 26, 2867–2873. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Levine, D.; Shen, J.; Gogarten, S.M.; Laurie, C.; Weir, B.S. A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics 2012, 28, 3326–3328. [Google Scholar] [CrossRef]
- Bresadola, L.; Link, V.; Buerkle, C.A.; Lexer, C.; Wegmann, D. Estimating and accounting for genotyping errors in RAD-seq experiments. Mol. Ecol. Resour. 2020, 20, 856–870. [Google Scholar] [CrossRef]
- Lischer, H.E.; Excoffier, L. PGDSpider: An automated data conversion tool for connecting population genetics and genomics programs. Bioinformatics 2012, 28, 298–299. [Google Scholar] [CrossRef]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef]
- Foll, M. BayeScan v2.1 User Manual. 2012. Available online: http://cmpg.unibe.ch/software/BayeScan/ (accessed on 30 June 2022).
- Foll, M.; Gaggiotti, O. A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: A Bayesian perspective. Genetics 2008, 180, 977–993. [Google Scholar] [CrossRef] [PubMed]
- Lotterhos, K.E.; Whitlock, M.C. The relative power of genome scans to detect local adaptation depends on sampling design and statistical method. Mol. Ecol. 2015, 24, 1031–1046. [Google Scholar] [CrossRef] [PubMed]
- Geweke, J.F. Evaluating the Accuracy of Sampling-Based Approaches to the Calculation of Posterior Moments (No. 148); Federal Reserve Bank of Minneapolis: St. Louis, MO, USA, 1991. [Google Scholar]
- Plummer, M.; Best, N.; Cowles, K.; Vines, K. CODA: Convergence diagnosis and output analysis for MCMC. R News 2006, 6, 7–11. [Google Scholar]
- Luu, K.; Bazin, E.; Blum, M.G. pcadapt: An R package to perform genome scans for selection based on principal component analysis. Mol. Ecol. Resour. 2017, 17, 67–77. [Google Scholar] [CrossRef]
- Whitlock, M.C.; Lotterhos, K.E. Reliable detection of loci responsible for local adaptation: Inference of a null model through trimming the distribution of FST. Am. Nat. 2015, 186 (Suppl. S1), S24–S36. [Google Scholar] [CrossRef] [PubMed]
- Privé, F.; Luu, K.; Vilhjálmsson, B.J.; Blum, M.G.B. Performing highly efficient genome scans for local adaptation with R package pcadapt version 4. Mol. Biol. Evol. 2020, 37, 2153–2154. [Google Scholar] [CrossRef]
- Weir, B.S.; Goudet, J. A unified characterization of population structure and relatedness. Genetics 2017, 206, 2085–2103. [Google Scholar] [CrossRef]
- Goudet, J. Hierfstat, a package for R to compute and test hierarchical F-statistics. Mol. Ecol. Notes 2005, 5, 184–186. [Google Scholar] [CrossRef]
- Goudet, J.; Jombart, T. Hierfstat: Estimation and Tests of Hierarchical F-Statistics. 2022. Available online: https://www.r-project.org (accessed on 15 November 2022).
- Archer, F.I.; Adams, P.E.; Schneiders, B.B. stratag: An r package for manipulating, summarizing and analysing population genetic data. Mol. Ecol. Resour. 2017, 17, 5–11. [Google Scholar] [CrossRef]
- Weir, B.S.; Cockerham, C.C. Estimating F-statistics for the analysis of population structure. Evolution 1984, 1, 1358–1370. [Google Scholar]
- Redon, R.; Ishikawa, S.; Fitch, K.R.; Feuk, L.; Perry, G.H.; Andrews, T.D.; Fiegler, H.; Shapero, M.H.; Carson, A.R.; Chen, W.; et al. Global variation in copy number in the human genome. Nature 2006, 444, 444–454. [Google Scholar] [CrossRef] [PubMed]
- Dennis, M.Y.; Harshman, L.; Nelson, B.J.; Penn, O.; Cantsilieris, S.; Huddleston, J.; Antonacci, F.; Penewit, K.; Denman, L.; Raja, A.; et al. The evolution and population diversity of human-specific segmental duplications. Nat. Ecol. Evol. 2017, 1, 0069. [Google Scholar] [CrossRef] [PubMed]
- Rinker, D.C.; Specian, N.K.; Zhao, S.; Gibbons, J.G. Polar bear evolution is marked by rapid changes in gene copy number in response to dietary shift. Proc. Natl. Acad. Sci. USA 2019, 116, 13446–13451. [Google Scholar] [CrossRef] [PubMed]
- Nei, M. Genetic distance between populations. Am. Nat. 1972, 106, 283–292. [Google Scholar] [CrossRef]
- Nei, M.; Takezaki, N. Estimation of genetic distances and phylogenetic trees from DNA analysis. In Proceedings of the 5th World Congress on Genetics Applied to Livestock Production, Guelph, ON, Canada, 7–12 August 1994; Volume 21, pp. 405–412. [Google Scholar]
- Pembleton, L.W.; Cogan, N.O.; Forster, J.W. StAMPP: An R package for calculation of genetic differentiation and structure of mixed-ploidy level populations. Mol. Ecol. Resour. 2013, 13, 946–952. [Google Scholar] [CrossRef] [PubMed]
- Paradis, E.; Schliep, K. ape 5.0: An environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 2019, 35, 526–528. [Google Scholar] [CrossRef]
- Jombart, T.; Devillard, S.; Balloux, F. Discriminant analysis of principal components: A new method for the analysis of genetically structured populations. BMC Genet. 2010, 11, 94. [Google Scholar] [CrossRef]
- Jombart, T.; Ahmed, I. adegenet 1.3-1: New tools for the analysis of genome-wide SNP data. Bioinformatics 2011, 27, 3070–3071. [Google Scholar] [CrossRef]
- Frichot, E.; François, O. LEA: An R package for landscape and ecological association studies. Methods Ecol. Evol. 2015, 6, 925–929. [Google Scholar] [CrossRef]
- Gain, C.; François, O. LEA 3: Factor models in population genetics and ecological genomics with R. Mol. Ecol. Resour. 2021, 21, 2738–2748. [Google Scholar] [CrossRef]
- Alexander, D.H.; Lange, K. Enhancements to the ADMIXTURE algorithm for individual ancestry estimation. BMC Bioinform. 2011, 12, 246. [Google Scholar] [CrossRef] [PubMed]
- Francis, R.M. pophelper: An R package and web app to analyse and visualize population structure. Mol. Ecol. Resour. 2017, 17, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Malinsky, M.; Trucchi, E.; Lawson, D.J.; Falush, D. RADpainter and fineRADstructure: Population inference from RADseq data. Mol. Biol. Evol. 2018, 35, 1284–1290. [Google Scholar] [CrossRef] [PubMed]
- Lawson, D.J.; Hellenthal, G.; Myers, S.; Falush, D. Inference of population structure using dense haplotype data. PLoS Genet. 2012, 8, e1002453. [Google Scholar] [CrossRef] [PubMed]
- Rellstab, C.; Gugerli, F.; Eckert, A.J.; Hancock, A.M.; Holderegger, R. A practical guide to environmental association analysis in landscape genomics. Mol. Ecol. 2015, 24, 4348–4370. [Google Scholar] [CrossRef]
- Riginos, C.; Crandall, E.D.; Liggins, L.; Bongaerts, P.; Treml, E.A. Navigating the currents of seascape genomics: How spatial analyses can augment population genomic studies. Curr. Zool. 2016, 62, 581–601. [Google Scholar] [CrossRef]
- Leempoel, K.; Duruz, S.; Rochat, E.; Widmer, I.; Orozco-terWengel, P.; Joost, S. Simple rules for an efficient use of geographic information systems in molecular ecology. Front. Ecol. Evol. 2017, 5, 33. [Google Scholar] [CrossRef]
- Riginos, C.; Liggins, L. Seascape genetics: Populations, individuals, and genes marooned and adrift. Geogr. Compass 2013, 7, 197–216. [Google Scholar] [CrossRef]
- Benestan, L.; Quinn, B.K.; Maaroufi, H.; Laporte, M.; Clark, F.K.; Greenwood, S.J.; Rochette, R.; Bernatchez, L. Seascape genomics provides evidence for thermal adaptation and current-mediated population structure in American lobster (Homarus americanus). Mol. Ecol. 2016, 25, 5073–5092. [Google Scholar] [CrossRef]
- Selkoe, K.; D’aloia, C.; Crandall, E.; Iacchei, M.; Liggins, L.; Puritz, J.; von der Heyden, S.; Toonen, R. A decade of seascape genetics: Contributions to basic and applied marine connectivity. Mar. Ecol. Prog. Ser. 2016, 554, 1–19. [Google Scholar] [CrossRef]
- Cayuela, H.; Rougemont, Q.; Laporte, M.; Mérot, C.; Normandeau, E.; Dorant, Y.; Tørresen, O.K.; Hoff, S.N.K.; Jentoft, S.; Sirois, P.; et al. Shared ancestral polymorphisms and chromosomal rearrangements as potential drivers of local adaptation in a marine fish. Mol. Ecol. 2020, 29, 2379–2398. [Google Scholar] [CrossRef] [PubMed]
- Hijmans, R.J. Raster: Geographic data analysis and modeling. R Package 2016, 734, 473. [Google Scholar]
- Barbosa, A.M. fuzzySim: Applying fuzzy logic to binary similarity indices in ecology. Methods Ecol. Evol. 2015, 6, 853–858. [Google Scholar] [CrossRef]
- Marquardt, D.W. Generalized inverses, ridge regression, biased linear estimation and nonlinear estimation. Technometrics 1970, 12, 591–612. [Google Scholar] [CrossRef]
- Mansfield, E.R.; Helms, B.P. Detecting multicollinearity. Am. Stat. 1982, 36, 158–160. [Google Scholar]
- Roever, C.; Raabe Luebke, K.; Ligges, U.; Szepannek, G.; Zentgraf, M.; Meyer, D. klaR: Classification and Visualization, R Package Version 0.6-15. 2023. Available online: https://cran.r-project.org/web/packages/klaR/index.html (accessed on 15 June 2023).
- Dray, S.; Legendre, P.; Peres-Neto, P.R. Spatial modelling: A comprehensive framework for principal coordinate analysis of neighbour matrices (PCNM). Ecol. Model. 2006, 196, 483–493. [Google Scholar] [CrossRef]
- Dray, S.; Pélissier, R.; Couteron, P.; Fortin, M.-J.; Legendre, P.; Peres-Neto, P.R.; Bellier, E.; Bivand, R.; Blanchet, F.G.; De Cáceres, M.; et al. Community ecology in the age of multivariate spatial analysis. Ecol. Monogr. 2012, 82, 257–275. [Google Scholar] [CrossRef]
- Peres-Neto, P.R.; Legendre, P. Estimating and controlling for spatial structure in the study of ecological communities. Glob. Ecol. Biogeogr. 2010, 19, 174–184. [Google Scholar] [CrossRef]
- Borcard, D.; Gillet, F.; Legendre, P.; Borcard, D.; Gillet, F.; Legendre, P. Spatial analysis of ecological data. In Numerical Ecology with R; Springer: New York, NY, USA, 2018; pp. 299–367. [Google Scholar] [CrossRef]
- Chambers, J.M. No. 1., Software for Data Analysis: Programming with R; Springer: New York, NY, USA, 2008; Volume 2. [Google Scholar]
- Pante, E.; Simon-Bouhet, B. marmap: A package for importing, plotting and analyzing bathymetric and topographic data in R. PLoS ONE 2013, 8, e73051. [Google Scholar] [CrossRef]
- Dray, S.; Bauman, D.; Blanchet, G.; Borcard, D.; Clappe, S.; Guénard, G.; Madi, N.H. Adespatial: Multivariate Multiscale Spatial Analysis. R Package 2022, 1–138. Available online: https://cran.r-project.org/src/contrib/Archive/adespatial/ (accessed on 20 August 2023).
- Dray, S.; Dufour, A.B. The ade4 Package: Implementing the duality diagram for ecologists. J. Stat. Softw. 2007, 22, 1–20. [Google Scholar] [CrossRef]
- Forester, B.R.; Lasky, J.R.; Wagner, H.H.; Urban, D.L. Comparing methods for detecting multilocus adaptation with multivariate genotype-environment associations. Mol. Ecol. 2018, 27, 2215–2233. [Google Scholar] [CrossRef] [PubMed]
- Caye, K.; Jumentier, B.; Lepeule, J.; François, O. LFMM 2: Fast and accurate inference of gene-environment associations in genome-wide studies. Mol. Biol. Evol. 2019, 36, 852–860. [Google Scholar] [CrossRef] [PubMed]
- Ahrens, C.W.; Rymer, P.D.; Stow, A.; Bragg, J.; Dillon, S.; Umbers, K.D.; Dudaniec, R.Y. The search for loci under selection: Trends, biases and progress. Mol. Ecol. 2018, 27, 1342–1356. [Google Scholar] [CrossRef]
- Waldvogel, A.M.; Schreiber, D.; Pfenninger, M.; Feldmeyer, B. Climate change genomics calls for standardized data reporting. Front. Ecol. Evol. 2020, 8, 242. [Google Scholar] [CrossRef]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Package ‘Vegan’. Community Ecology Package, Version. 2019. Available online: https://CRAN.R-project.org/package=vegan (accessed on 25 November 2022).
- Macarthur, R.H. On the relative abundance of bird species. Proc. Natl. Acad. Sci. USA 1957, 43, 293–295. [Google Scholar] [CrossRef]
- King, J.R.; Jackson, D.A. Variable selection in large environmental data sets using principal components analysis. Environmetrics Off. J. Int. Environmetrics Soc. 1999, 10, 67–77. [Google Scholar] [CrossRef]
- Storey, J.D. The positive false discovery rate: A Bayesian interpretation and the q-value. Ann. Stat. 2003, 31, 2013–2035. [Google Scholar] [CrossRef]
- Capblancq, T.; Luu, K.; Blum, M.G.B.; Bazin, E. Evaluation of redundancy analysis to identify signatures of local adaptation. Mol. Ecol. Resour. 2018, 18, 1223–1233. [Google Scholar] [CrossRef]
- Cingolani, P. Variant annotation and functional prediction: SnpEff. In Variant Calling: Methods and Protocols; Springer: New York, NY, USA, 2018; pp. 289–314. [Google Scholar]
- Brodie, A.; Azaria, J.R.; Ofran, Y. How far from the SNP may the causative genes be? Nucleic Acids Res. 2016, 44, 6046–6054. [Google Scholar] [CrossRef]
- Visel, A.; Rubin, E.M.; Pennacchio, L.A. Genomic views of distant-acting enhancers. Nature 2009, 461, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Selmoni, O.; Rochat, E.; Lecellier, G.; Berteaux-Lecellier, V.; Joost, S. Seascape genomics as a new tool to empower coral reef conservation strategies: An example on north-western Pacific Acropora digitifera. Evol. Appl. 2020, 13, 1923–1938. [Google Scholar] [CrossRef] [PubMed]
- Madden, T.; Coulouris, G. BLAST Command Line Applications User Manual BLAST Command Line Applications User Manual-BLAST; National Center for Biotechnology Information: Bethesda, MD, USA, 2008. Available online: http://www.ncbi.nlm.nih.gov/books/NBK279690/ (accessed on 15 March 2023).
- Boeckmann, B.; Bairoch, A.; Apweiler, R.; Blatter, M.C.; Estreicher, A.; Gasteiger, E.; Martin, M.J.; Michoud, K.; O’Donovan, C.; Phan, I.; et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 2003, 31, 365–370. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef] [PubMed]
- Charlesworth, B. Fundamental concepts in genetics: Effective population size and patterns of molecular evolution and variation. Nat. Rev. Genet. 2009, 10, 195–205. [Google Scholar] [CrossRef]
- Greene, J.S.; Brown, M.; Dobosiewicz, M.; Ishida, I.G.; Macosko, E.Z.; Zhang, X.; Butcher, R.A.; Cline, D.J.; McGrath, P.T.; Bargmann, C.I. Balancing selection shapes density-dependent foraging behaviour. Nature 2016, 539, 254–258. [Google Scholar] [CrossRef]
- Nettle, D. The evolution of personality variation in humans and other animals. Am. Psychol. 2006, 61, 622–631. [Google Scholar] [CrossRef]
- Penke, L.; Denissen, J.J.; Miller, G.F. The evolutionary genetics of personality. Eur. J. Personal. 2007, 21, 549–587. [Google Scholar] [CrossRef]
- Christie, M.R.; McNickle, G.G.; French, R.A.; Blouin, M.S. Life history variation is maintained by fitness trade-offs and negative frequency-dependent selection. Proc. Natl. Acad. Sci. USA 2018, 115, 4441–4446. [Google Scholar] [CrossRef]
- Cavedon, M.; Goubili, C.; Heppenheimer, E.; Vonholdt, B.; Mariani, S.; Hebblewhite, M.; Hegel, T.; Hervieux, D.; Serrouya, R.; Steenweg, R.; et al. Genomics, environment and balancing selection in behaviourally bimodal populations: The caribou case. Mol. Ecol. 2019, 28, 1946–1963. [Google Scholar] [CrossRef]
- Carrier, A.; Prunier, J.; Poisson, W.; Trottier-Lavoie, M.; Gilbert, I.; Cavedon, M.; Pokharel, K.; Kantanen, J.; Musiani, M.; Côté, S.D.; et al. Design and validation of a 63K genome-wide SNP-genotyping platform for caribou/reindeer (Rangifer tarandus). BMC Genom. 2022, 23, 687. [Google Scholar] [CrossRef] [PubMed]
- Halvorsen, K.T.; Larsen, T.; Sørdalen, T.K.; Vøllestad, L.A.; Knutsen, H.; Olsen, E.M. Impact of harvesting cleaner fish for salmonid aquaculture assessed from replicated coastal marine protected areas. Mar. Biol. Res. 2017, 13, 359–369. [Google Scholar] [CrossRef]
- Imsland, A.K.; Reynolds, P.; Nytrø, A.V.; Eliassen, G.; Hangstad, T.A.; Jónsdóttir, Ó.D.; Emaus, P.-A.; Elvegård, T.A.; Lemmens, S.C.; Rydland, R.; et al. Effects of lumpfish size on foraging behaviour and co-existence with sea lice infected Atlantic salmon in sea cages. Aquaculture 2016, 465, 19–27. [Google Scholar] [CrossRef]
- Imsland, A.K.; Reynolds, P.; Eliassen, G.; Mortensen, A.; Hansen, Ø.J.; Puvanendran, V.; Hangstad, T.A.; Jónsdóttir, Ó.D.; Emaus, P.A.; Elvegård, T.A.; et al. Is cleaning behaviour in lumpfish (Cyclopterus lumpus) parentally controlled? Aquaculture 2016, 459, 156–165. [Google Scholar] [CrossRef]
- Whittaker, B.A.; Consuegra, S.; de Leaniz, C.G. Personality profiling may help select better cleaner fish for sea-lice control in salmon farming. Appl. Anim. Behav. Sci. 2021, 243, 105459. [Google Scholar] [CrossRef]
- Darwin, C. On the Origin of Species by Means of Natural Selection, or the Preservation of Favoured Races in the Struggle for Life; John Murray: London, UK, 1859. [Google Scholar] [CrossRef]
- Hedrick, P.W.; Ginevan, M.E.; Ewing, E.P. Genetic polymorphism in heterogeneous environments. Annu. Rev. Ecol. Syst. 1976, 7, 1–32. [Google Scholar] [CrossRef]
- Palumbi, S.R. Genetic divergence, reproductive isolation, and marine speciation. Annu. Rev. Ecol. Syst. 1994, 25, 547–572. [Google Scholar] [CrossRef]
- Selkoe, K.A.; Henzler, C.M.; Gaines, S.D. Seascape genetics and the spatial ecology of marine populations. Fish Fish. 2008, 9, 363–377. [Google Scholar] [CrossRef]
- Coscia, I.; Wilmes, S.B.; Ironside, J.E.; Goward-Brown, A.; O’Dea, E.; Malham, S.K.; McDevitt, A.D.; Robins, P.E. Fine-scale seascape genomics of an exploited marine species, the common cockle Cerastoderma edule, using a multimodelling approach. Evol. Appl. 2020, 13, 1854–1867. [Google Scholar] [CrossRef]
- Huang, J.; Li, Y.; Liu, Z.; Kang, Y.; Wang, J. Transcriptomic responses to heat stress in rainbow trout Oncorhynchus mykiss head kidney. Fish Shellfish Immunol. 2018, 82, 32–40. [Google Scholar] [CrossRef]
- Huang, Z.; Ma, A.; Yang, S.; Liu, X.; Zhao, T.; Zhang, J.; Wang, X.A.; Sun, Z.; Liu, Z.; Xu, R. Transcriptome analysis and weighted gene co-expression network reveals potential genes responses to heat stress in turbot Scophthalmus maximus. Comp. Biochem. Physiol. Part D Genom. Proteom. 2020, 33, 100632. [Google Scholar] [CrossRef] [PubMed]
- Uthicke, S.; Deshpande, N.P.; Liddy, M.; Patel, F.; Lamare, M.; Wilkins, M.R. Little evidence of adaptation potential to ocean acidification in sea urchins living in “Future Ocean” conditions at a CO2 vent. Ecol. Evol. 2019, 9, 10004–10016. [Google Scholar] [CrossRef] [PubMed]
- Petit-Marty, N.; Nagelkerken, I.; Connell, S.D.; Schunter, C. Natural CO2 seeps reveal adaptive potential to ocean acidification in fish. Evol. Appl. 2021, 14, 1794–1806. [Google Scholar] [CrossRef] [PubMed]
- De Wit, P.; Palumbi, S.R. Transcriptome-wide polymorphisms of red abalone (Haliotis rufescens) reveal patterns of gene flow and local adaptation. Mol. Ecol. 2013, 22, 2884–2897. [Google Scholar] [CrossRef] [PubMed]
- Hammerschmidt, M.; Brook, A.; McMahon, A.P. The world according to hedgehog. Trends Genet. 1997, 13, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Chuang, P.T.; McMahon, A.P. Vertebrate Hedgehog signalling modulated by induction of a Hedgehog-binding protein. Nature 1999, 397, 617–621. [Google Scholar] [CrossRef]
- Katoh, M.; Katoh, M. WNT signaling pathway and stem cell signaling network. Clin. Cancer Res. 2007, 13, 4042–4045. [Google Scholar] [CrossRef]
- Chen, R.E.; Thorner, J. Function and regulation in MAPK signaling pathways: Lessons learned from the yeast Saccharomyces cerevisiae. Biochim. Biophys. Acta 2007, 1773, 1311–1340. [Google Scholar] [CrossRef]
- Rehwinkel, J.; Gack, M.U. RIG-I-like receptors: Their regulation and roles in RNA sensing. Nat. Rev. Immunol. 2020, 20, 537–551. [Google Scholar] [CrossRef]












| NMA | NRG | NAU | NRA | NAV | NEK | NNA | NSA | NHE | NAL | GRE | CAN | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NMA | 0.111 (0.067–0.156) | 0.345 (0.232–0.458) | 0.134 (0.057–0.212) | 0.089 (0.037–0.141) | 0.182 (0.11–0.254) | 0.159 (0.065–0.252) | 0.219 (0.123–0.314) | 0.095 (0.056–0.134) | 0.046 (0.019–0.072) | 0.147 (0.082–0.213) | 0.317 (0.213–0.421) | |
| NRG | 0.019 (0.017–0.021) | 0.127 (0.065–0.188) | 0.19 (0.098–0.281) | 0.139 (0.074–0.203) | 0.243 (0.148–0.338) | 0.301 (0.193–0.408) | 0.116 (0.065–0.168) | 0.078 (0.042–0.115) | 0.057 (0.033–0.08) | 0.219 (0.13–0.309) | 0.163 (0.099–0.227) | |
| NAU | 0.009 (0.006–0.012) | 0.032 (0.028–0.035) | 0 (0–0) | 0.2 (0.105–0.295) | 0.271 (0.18–0.363) | 0.169 (0.062–0.277) | 0.272 (0.168–0.377) | 0.127 (0.065–0.189) | 0.054 (0.02–0.088) | 0.103 (0.039–0.166) | 0.276 (0.179–0.374) | |
| NRA | 0.004 (0.002–0.007) | 0.032 (0.028–0.035) | 0.01 (0.006–0.015) | 0.174 (0.107–0.242) | 0.248 (0.148–0.349) | 0.325 (0.212–0.437) | 0.181 (0.087–0.275) | 0.238 (0.151–0.325) | 0.077 (0.035–0.119) | 0.121 (0.048–0.195) | 0.287 (0.189–0.385) | |
| NAV | 0.007 (0.005–0.009) | 0.029 (0.026–0.031) | 0.015 (0.012–0.018) | 0.006 (0.003–0.009) | 0.185 (0.084–0.286) | 0.198 (0.125–0.271) | 0.154 (0.068–0.24) | 0.207 (0.132–0.283) | 0.115 (0.058–0.172) | 0.138 (0.061–0.216) | 0.226 (0.137–0.315) | |
| NEK | 0.003 (0–0.005) | 0.03 (0.027–0.034) | 0.008 (0.004–0.013) | 0.005 (0.001–0.009) | 0.001 (−0.002–0.005) | 0.191 (0.083–0.3) | 0.258 (0.144–0.372) | 0.123 (0.07–0.176) | 0.1 (0.039–0.161) | 0.126 (0.054–0.198) | 0.313 (0.198–0.428) | |
| NNA | 0.004 (0.003–0.006) | 0.027 (0.025–0.03) | 0.011 (0.009–0.014) | 0.002 (−0.001–0.005) | 0.006 (0.004–0.008) | −0.002 (−0.004–0.001) | 0.076 (0.037–0.114) | 0.087 (0.051–0.123) | 0.088 (0.05–0.126) | 0.087 (0.046–0.128) | 0.308 (0.208–0.409) | |
| NSA | 0.006 (0.004–0.008) | 0.03 (0.027–0.033) | 0.016 (0.013–0.019) | 0.008 (0.005–0.011) | 0.004 (0.002–0.005) | 0.005 (0.002–0.008) | 0.005 (0.003–0.006) | 0.27 (0.162–0.378) | 0.094 (0.049–0.138) | 0.057 (0.013–0.101) | 0.331 (0.22–0.442) | |
| NHE | 0.005 (0.004–0.007) | 0.026 (0.024–0.029) | 0.014 (0.011–0.017) | 0.003 (0.001–0.006) | 0.005 (0.003–0.007) | 0.001 (−0.002–0.003) | 0.002 (0–0.003) | 0.003 (0.001–0.004) | 0.098 (0.064–0.131) | 0.113 (0.062–0.163) | 0.158 (0.094–0.222) | |
| NAL | 0.008 (0.007–0.01) | 0.031 (0.028–0.033) | 0.015 (0.012–0.018) | 0.003 (0.001–0.006) | 0.01 (0.008–0.012) | 0.002 (−0.001–0.005) | 0.007 (0.005–0.008) | 0.008 (0.007–0.01) | 0.006 (0.005–0.008) | 0.061 (0.036–0.086) | 0.179 (0.12–0.238) | |
| GRE | 0.118 (0.114–0.122) | 0.141 (0.137–0.146) | 0.134 (0.129–0.138) | 0.132 (0.126–0.137) | 0.127 (0.122–0.131) | 0.124 (0.118–0.129) | 0.122 (0.118–0.126) | 0.126 (0.122–0.131) | 0.123 (0.119–0.127) | 0.118 (0.114–0.122) | 0.048 (0.023–0.074) | |
| CAN | 0.153 (0.148–0.159) | 0.182 (0.176–0.187) | 0.17 (0.164–0.176) | 0.176 (0.169–0.182) | 0.165 (0.159–0.171) | 0.165 (0.158–0.171) | 0.156 (0.151–0.162) | 0.163 (0.158–0.168) | 0.157 (0.152–0.163) | 0.152 (0.147–0.157) | 0.068 (0.065–0.071) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maduna, S.N.; Jónsdóttir, Ó.D.B.; Imsland, A.K.D.; Gíslason, D.; Reynolds, P.; Kapari, L.; Hangstad, T.A.; Meier, K.; Hagen, S.B. Genomic Signatures of Local Adaptation under High Gene Flow in Lumpfish—Implications for Broodstock Provenance Sourcing and Larval Production. Genes 2023, 14, 1870. https://doi.org/10.3390/genes14101870
Maduna SN, Jónsdóttir ÓDB, Imsland AKD, Gíslason D, Reynolds P, Kapari L, Hangstad TA, Meier K, Hagen SB. Genomic Signatures of Local Adaptation under High Gene Flow in Lumpfish—Implications for Broodstock Provenance Sourcing and Larval Production. Genes. 2023; 14(10):1870. https://doi.org/10.3390/genes14101870
Chicago/Turabian StyleMaduna, Simo Njabulo, Ólöf Dóra Bartels Jónsdóttir, Albert Kjartan Dagbjartarson Imsland, Davíð Gíslason, Patrick Reynolds, Lauri Kapari, Thor Arne Hangstad, Kristian Meier, and Snorre B. Hagen. 2023. "Genomic Signatures of Local Adaptation under High Gene Flow in Lumpfish—Implications for Broodstock Provenance Sourcing and Larval Production" Genes 14, no. 10: 1870. https://doi.org/10.3390/genes14101870