
Current Status and Prospects of the Single-Cell Sequencing Technologies for Revealing the Pathogenesis of Pregnancy-Associated Disorders
 and
and 
Abstract
:1. Introduction
2. scRNA-seq: Laboratory Technologies and Bioinformatic Data Analysis Instruments
2.1. Laboratory Protocols
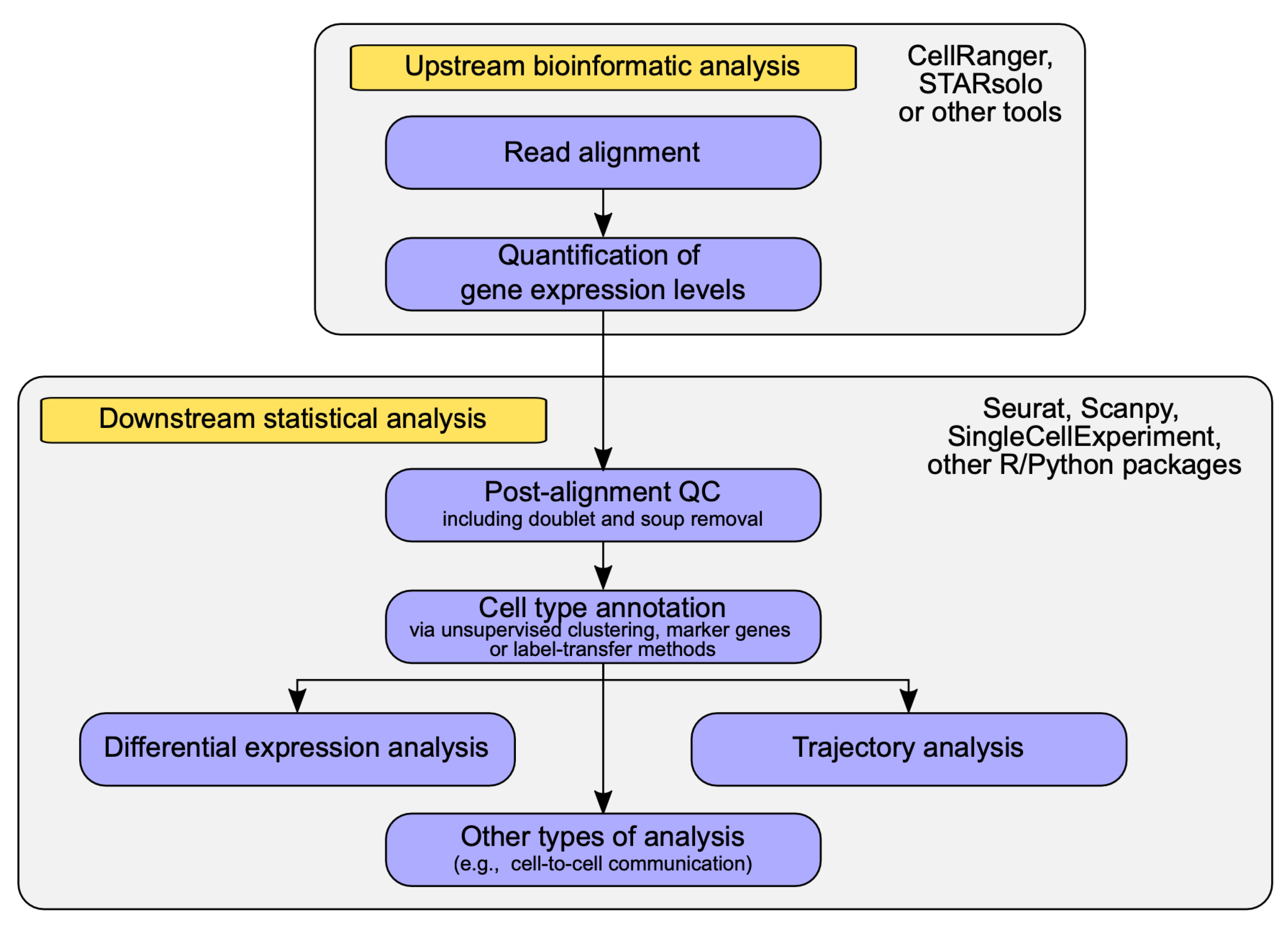
2.2. Data Analysis Methods in scRNA-seq
3. The Recent Discoveries in the scRNA-seq Studies of Human Pregnancy
3.1. Results of the scRNA-seq Studies of Normal Pregnancy Conditions
3.2. Results of the scRNA-seq Studies of Pregnancy Complications and Pregnancy-Associated Diseases
3.2.1. Hyperglycemia in Pregnancy
3.2.2. Preeclampsia
3.2.3. Preterm Labor
3.2.4. Recurrent Pregnancy Loss
3.2.5. Conditions Associated with an Increased Risk of Pregnancy Complications
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A Revolutionary Tool for Transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Yong, H.E.J.; Chan, S.-Y. Current Approaches and Developments in Transcript Profiling of the Human Placenta. Hum. Reprod. Update 2020, 26, 799–840. [Google Scholar] [CrossRef] [PubMed]
- Cox, B.; Leavey, K.; Nosi, U.; Wong, F.; Kingdom, J. Placental Transcriptome in Development and Pathology: Expression, Function, and Methods of Analysis. Am. J. Obstet. Gynecol. 2015, 213, S138–S151. [Google Scholar] [CrossRef] [PubMed]
- Tong, J.; Zhao, W.; Lv, H.; Li, W.-P.; Chen, Z.-J.; Zhang, C. Transcriptomic Profiling in Human Decidua of Severe Preeclampsia Detected by RNA Sequencing. J. Cell. Biochem. 2018, 119, 607–615. [Google Scholar] [CrossRef]
- Zhao, Y.-H.; Wang, D.-P.; Zhang, L.-L.; Zhang, F.; Wang, D.-M.; Zhang, W.-Y. Genomic Expression Profiles of Blood and Placenta Reveal Significant Immune-Related Pathways and Categories in Chinese Women with Gestational Diabetes Mellitus. Diabet. Med. 2011, 28, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Binder, A.M.; LaRocca, J.; Lesseur, C.; Marsit, C.J.; Michels, K.B. Epigenome-Wide and Transcriptome-Wide Analyses Reveal Gestational Diabetes Is Associated with Alterations in the Human Leukocyte Antigen Complex. Clin. Epigenet. 2015, 7, 79. [Google Scholar] [CrossRef] [Green Version]
- Mouillet, J.-F.; Ouyang, Y.; Coyne, C.B.; Sadovsky, Y. MicroRNAs in Placental Health and Disease. Am. J. Obstet. Gynecol. 2015, 213, S163–S172. [Google Scholar] [CrossRef] [Green Version]
- Vashukova, E.S.; Kozyulina, P.Y.; Illarionov, R.A.; Yurkina, N.O.; Pachuliia, O.V.; Butenko, M.G.; Postnikova, T.B.; Ivanova, L.A.; Eremeeva, D.R.; Zainulina, M.S.; et al. High-Throughput Sequencing of Circulating MicroRNAs in Plasma and Serum during Pregnancy Progression. Life 2021, 11, 1055. [Google Scholar] [CrossRef]
- Gu, Y.; Sun, J.; Groome, L.J.; Wang, Y. Differential MiRNA Expression Profiles between the First and Third Trimester Human Placentas. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E836–E843. [Google Scholar] [CrossRef] [Green Version]
- Tonyan, Z.N.; Nasykhova, Y.A.; Danilova, M.M.; Barbitoff, Y.A.; Changalidi, A.I.; Mikhailova, A.A.; Glotov, A.S. Overview of Transcriptomic Research on Type 2 Diabetes: Challenges and Perspectives. Genes 2022, 13, 1176. [Google Scholar] [CrossRef]
- Hu, J.; Tang, T.; Zeng, Z.; Wu, J.; Tan, X.; Yan, J. The Expression of Small RNAs in Exosomes of Follicular Fluid Altered in Human Polycystic Ovarian Syndrome. PeerJ 2020, 8, e8640. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Li, S.-P.; Zhang, T.; Yu, B.; Zhang, J.; Ding, H.-G.; Ye, F.-J.; Yuan, H.; Ma, Y.-Y.; Pan, H.-T.; et al. High Throughput MicroRNAs Sequencing Profile of Serum Exosomes in Women with and without Polycystic Ovarian Syndrome. PeerJ 2021, 9, e10998. [Google Scholar] [CrossRef]
- PREGMIR|MiRNA Database. Available online: https://pregmir.ott.ru/ (accessed on 11 January 2023).
- Gong, S.; Gaccioli, F.; Dopierala, J.; Sovio, U.; Cook, E.; Volders, P.-J.; Martens, L.; Kirk, P.D.W.; Richardson, S.; Smith, G.C.S.; et al. The RNA Landscape of the Human Placenta in Health and Disease. Nat. Commun. 2021, 12, 2639. [Google Scholar] [CrossRef] [PubMed]
- Ding, R.; Guo, F.; Zhang, Y.; Liu, X.-M.; Xiang, Y.-Q.; Zhang, C.; Liu, Z.-W.; Sheng, J.-Z.; Huang, H.-F.; Zhang, J.-Y.; et al. Integrated Transcriptome Sequencing Analysis Reveals Role of MiR-138-5p/ TBL1X in Placenta from Gestational Diabetes Mellitus. Cell. Physiol. Biochem. 2018, 51, 630–646. [Google Scholar] [CrossRef] [PubMed]
- Xu, N.; Zhou, X.; Shi, W.; Ye, M.; Cao, X.; Chen, S.; Xu, C. Integrative Analysis of Circulating MicroRNAs and the Placental Transcriptome in Recurrent Pregnancy Loss. Front. Physiol. 2022, 13, 893744. [Google Scholar] [CrossRef]
- Nieto, P.; Elosua-Bayes, M.; Trincado, J.L.; Marchese, D.; Massoni-Badosa, R.; Salvany, M.; Henriques, A.; Nieto, J.; Aguilar-Fernández, S.; Mereu, E.; et al. A Single-Cell Tumor Immune Atlas for Precision Oncology. Genome Res. 2021, 31, 1913–1926. [Google Scholar] [CrossRef]
- Lake, B.B.; Ai, R.; Kaeser, G.E.; Salathia, N.S.; Yung, Y.C.; Liu, R.; Wildberg, A.; Gao, D.; Fung, H.-L.; Chen, S.; et al. Neuronal Subtypes and Diversity Revealed by Single-Nucleus RNA Sequencing of the Human Brain. Science 2016, 352, 1586–1590. [Google Scholar] [CrossRef] [Green Version]
- Papalexi, E.; Satija, R. Single-Cell RNA Sequencing to Explore Immune Cell Heterogeneity. Nat. Rev. Immunol. 2018, 18, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Huang, Q.; Liu, Y.; Garmire, L.X. Single Cell Transcriptome Research in Human Placenta. Reproduction 2020, 160, R155–R167. [Google Scholar] [CrossRef]
- Vento-Tormo, R.; Efremova, M.; Botting, R.A.; Turco, M.Y.; Vento-Tormo, M.; Meyer, K.B.; Park, J.-E.; Stephenson, E.; Polański, K.; Goncalves, A.; et al. Single-Cell Reconstruction of the Early Maternal–Fetal Interface in Humans. Nature 2018, 563, 347–353. [Google Scholar] [CrossRef] [Green Version]
- Tang, F.; Barbacioru, C.; Wang, Y.; Nordman, E.; Lee, C.; Xu, N.; Wang, X.; Bodeau, J.; Tuch, B.B.; Siddiqui, A.; et al. MRNA-Seq Whole-Transcriptome Analysis of a Single Cell. Nat. Methods 2009, 6, 377–382. [Google Scholar] [CrossRef]
- Macosko, E.Z.; Basu, A.; Satija, R.; Nemesh, J.; Shekhar, K.; Goldman, M.; Tirosh, I.; Bialas, A.R.; Kamitaki, N.; Martersteck, E.M.; et al. Highly Parallel Genome-Wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 2015, 161, 1202–1214. [Google Scholar] [CrossRef] [Green Version]
- Shaffer, S.M.; Dunagin, M.C.; Torborg, S.R.; Torre, E.A.; Emert, B.; Krepler, C.; Beqiri, M.; Sproesser, K.; Brafford, P.A.; Xiao, M.; et al. Rare Cell Variability and Drug-Induced Reprogramming as a Mode of Cancer Drug Resistance. Nature 2017, 546, 431–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chavkin, N.W.; Hirschi, K.K. Single Cell Analysis in Vascular Biology. Front. Cardiovasc. Med. 2020, 7, 42. [Google Scholar] [CrossRef] [PubMed]
- Picelli, S.; Björklund, Å.K.; Faridani, O.R.; Sagasser, S.; Winberg, G.; Sandberg, R. Smart-Seq2 for Sensitive Full-Length Transcriptome Profiling in Single Cells. Nat. Methods 2013, 10, 1096–1098. [Google Scholar] [CrossRef]
- Fan, X.; Zhang, X.; Wu, X.; Guo, H.; Hu, Y.; Tang, F.; Huang, Y. Single-Cell RNA-Seq Transcriptome Analysis of Linear and Circular RNAs in Mouse Preimplantation Embryos. Genome Biol. 2015, 16, 148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheng, K.; Cao, W.; Niu, Y.; Deng, Q.; Zong, C. Effective Detection of Variation in Single-Cell Transcriptomes Using MATQ-Seq. Nat. Methods 2017, 14, 267–270. [Google Scholar] [CrossRef]
- Gierahn, T.M.; Wadsworth, M.H.; Hughes, T.K.; Bryson, B.D.; Butler, A.; Satija, R.; Fortune, S.; Love, J.C.; Shalek, A.K. Seq-Well: A Portable, Low-Cost Platform for High-Throughput Single-Cell RNA-Seq of Low-Input Samples. Nat. Methods 2017, 14, 395–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, G.X.Y.; Terry, J.M.; Belgrader, P.; Ryvkin, P.; Bent, Z.W.; Wilson, R.; Ziraldo, S.B.; Wheeler, T.D.; McDermott, G.P.; Zhu, J.; et al. Massively Parallel Digital Transcriptional Profiling of Single Cells. Nat. Commun. 2017, 8, 14049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habib, N.; Avraham-Davidi, I.; Basu, A.; Burks, T.; Shekhar, K.; Hofree, M.; Choudhury, S.R.; Aguet, F.; Gelfand, E.; Ardlie, K.; et al. Massively-Parallel Single Nucleus RNA-Seq with DroNc-Seq. Nat. Methods 2017, 14, 955–958. [Google Scholar] [CrossRef] [Green Version]
- Islam, S.; Kjällquist, U.; Moliner, A.; Zajac, P.; Fan, J.-B.; Lönnerberg, P.; Linnarsson, S. Highly Multiplexed and Strand-Specific Single-Cell RNA 5′ End Sequencing. Nat. Protoc. 2012, 7, 813–828. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Ning, B.; Shi, T. Single-Cell RNA-Seq Technologies and Related Computational Data Analysis. Front. Genet. 2019, 10, 317. [Google Scholar] [CrossRef]
- Shum, E.Y.; Walczak, E.M.; Chang, C.; Christina Fan, H. Quantitation of MRNA Transcripts and Proteins Using the BD RhapsodyTM Single-Cell Analysis System. In Single Molecule and Single Cell Sequencing; Advances in Experimental Medicine and, Biology; Suzuki, Y., Ed.; Springer: Singapore, 2019; pp. 63–79. ISBN 9789811360374. [Google Scholar]
- Goldstein, L.D.; Chen, Y.-J.J.; Dunne, J.; Mir, A.; Hubschle, H.; Guillory, J.; Yuan, W.; Zhang, J.; Stinson, J.; Jaiswal, B.; et al. Massively Parallel Nanowell-Based Single-Cell Gene Expression Profiling. BMC Genom. 2017, 18, 519. [Google Scholar] [CrossRef] [Green Version]
- Lafzi, A.; Moutinho, C.; Picelli, S.; Heyn, H. Tutorial: Guidelines for the Experimental Design of Single-Cell RNA Sequencing Studies. Nat. Protoc. 2018, 13, 2742–2757. [Google Scholar] [CrossRef] [Green Version]
- Kashima, Y.; Sakamoto, Y.; Kaneko, K.; Seki, M.; Suzuki, Y.; Suzuki, A. Single-Cell Sequencing Techniques from Individual to Multiomics Analyses. Exp. Mol. Med. 2020, 52, 1419–1427. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A Fast Spliced Aligner with Low Memory Requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Gingeras, T.R. Mapping RNA-Seq Reads with STAR. Curr. Protoc. Bioinform. 2015, 51, 11.14.1–11.14.19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate Transcript Quantification from RNA-Seq Data with or without a Reference Genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Kaminow, B.; Yunusov, D.; Dobin, A. STARsolo: Accurate, Fast and Versatile Mapping/Quantification of Single-Cell and Single-Nucleus RNA-Seq Data. bioRxiv 2021. [Google Scholar] [CrossRef]
- Melsted, P.; Booeshaghi, A.S.; Liu, L.; Gao, F.; Lu, L.; Min, K.H.J.; da Veiga Beltrame, E.; Hjörleifsson, K.E.; Gehring, J.; Pachter, L. Modular, Efficient and Constant-Memory Single-Cell RNA-Seq Preprocessing. Nat. Biotechnol. 2021, 39, 813–818. [Google Scholar] [CrossRef]
- Srivastava, A.; Malik, L.; Smith, T.; Sudbery, I.; Patro, R. Alevin Efficiently Estimates Accurate Gene Abundances from DscRNA-Seq Data. Genome Biol. 2019, 20, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satija, R.; Farrell, J.A.; Gennert, D.; Schier, A.F.; Regev, A. Spatial Reconstruction of Single-Cell Gene Expression. Nat. Biotechnol. 2015, 33, 495–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butler, A.; Hoffman, P.; Smibert, P.; Papalexi, E.; Satija, R. Integrating Single-Cell Transcriptomic Data across Different Conditions, Technologies, and Species. Nat. Biotechnol. 2018, 36, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck, W.M.; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive Integration of Single-Cell Data. Cell 2019, 177, 1888–1902.e21. [Google Scholar] [CrossRef]
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M.; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M.; et al. Integrated Analysis of Multimodal Single-Cell Data. Cell 2021, 184, 3573–3587.e29. [Google Scholar] [CrossRef]
- Amezquita, R.A.; Lun, A.T.L.; Becht, E.; Carey, V.J.; Carpp, L.N.; Geistlinger, L.; Marini, F.; Rue-Albrecht, K.; Risso, D.; Soneson, C.; et al. Orchestrating Single-Cell Analysis with Bioconductor. Nat. Methods 2020, 17, 137–145. [Google Scholar] [CrossRef]
- Wolf, F.A.; Angerer, P.; Theis, F.J. SCANPY: Large-Scale Single-Cell Gene Expression Data Analysis. Genome Biol. 2018, 19, 15. [Google Scholar] [CrossRef] [Green Version]
- Xi, N.M.; Li, J.J. Benchmarking Computational Doublet-Detection Methods for Single-Cell RNA Sequencing Data. Cell. Syst. 2021, 12, 176–194.e6. [Google Scholar] [CrossRef]
- Yang, S.; Corbett, S.E.; Koga, Y.; Wang, Z.; Johnson, W.E.; Yajima, M.; Campbell, J.D. Decontamination of Ambient RNA in Single-Cell RNA-Seq with DecontX. Genome Biol. 2020, 21, 57. [Google Scholar] [CrossRef]
- Svensson, V. Droplet ScRNA-Seq Is Not Zero-Inflated. Nat. Biotechnol. 2020, 38, 147–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McInnes, L.; Healy, J.; Melville, J. UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. arXiv 2018, arXiv:1802.03426. [Google Scholar]
- Kobak, D.; Berens, P. The Art of Using T-SNE for Single-Cell Transcriptomics. Nat. Commun. 2019, 10, 5416. [Google Scholar] [CrossRef] [Green Version]
- Ianevski, A.; Giri, A.K.; Aittokallio, T. Fully-Automated and Ultra-Fast Cell-Type Identification Using Specific Marker Combinations from Single-Cell Transcriptomic Data. Nat. Commun. 2022, 13, 1246. [Google Scholar] [CrossRef] [PubMed]
- Domínguez Conde, C.; Xu, C.; Jarvis, L.B.; Rainbow, D.B.; Wells, S.B.; Gomes, T.; Howlett, S.K.; Suchanek, O.; Polanski, K.; King, H.W.; et al. Cross-Tissue Immune Cell Analysis Reveals Tissue-Specific Features in Humans. Science 2022, 376, eabl5197. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Cao, Y.; Yang, J.Y.H.; Yang, P. Benchmarking Clustering Algorithms on Estimating the Number of Cell Types from Single-Cell RNA-Sequencing Data. Genome Biol. 2022, 23, 49. [Google Scholar] [CrossRef] [PubMed]
- Saelens, W.; Cannoodt, R.; Todorov, H.; Saeys, Y. A Comparison of Single-Cell Trajectory Inference Methods. Nat. Biotechnol. 2019, 37, 547–554. [Google Scholar] [CrossRef]
- Dimitrov, D.; Türei, D.; Garrido-Rodriguez, M.; Burmedi, P.L.; Nagai, J.S.; Boys, C.; Ramirez Flores, R.O.; Kim, H.; Szalai, B.; Costa, I.G.; et al. Comparison of Methods and Resources for Cell-Cell Communication Inference from Single-Cell RNA-Seq Data. Nat. Commun. 2022, 13, 3224. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Fan, X.; Wang, R.; Lu, X.; Dang, Y.-L.; Wang, H.; Lin, H.-Y.; Zhu, C.; Ge, H.; Cross, J.C.; et al. Single-Cell RNA-Seq Reveals the Diversity of Trophoblast Subtypes and Patterns of Differentiation in the Human Placenta. Cell. Res. 2018, 28, 819–832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suryawanshi, H.; Morozov, P.; Straus, A.; Sahasrabudhe, N.; Max, K.E.A.; Garzia, A.; Kustagi, M.; Tuschl, T.; Williams, Z. A Single-Cell Survey of the Human First-Trimester Placenta and Decidua. Sci. Adv. 2018, 4, eaau4788. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Li, J.; Wang, S.; Deng, Q.; An, Y.; Xing, Y.; Dai, X.; Li, Z.; Ma, Q.; Wang, K.; et al. Single-Cell Transcriptional Profiling Reveals Cellular and Molecular Divergence in Human Maternal-Fetal Interface. Sci. Rep. 2022, 12, 10892. [Google Scholar] [CrossRef]
- Li, H.; Peng, H.; Hong, W.; Wei, Y.; Tian, H.; Huang, X.; Jia, L.; Zheng, J.; Duan, T.; He, Q.; et al. Human Placental Endothelial Cell and Trophoblast Heterogeneity and Differentiation Revealed by Single-Cell RNA Sequencing. Cells 2022, 12, 87. [Google Scholar] [CrossRef]
- Pique-Regi, R.; Romero, R.; Garcia-Flores, V.; Peyvandipour, A.; Tarca, A.L.; Pusod, E.; Galaz, J.; Miller, D.; Bhatti, G.; Para, R.; et al. A Single-Cell Atlas of the Myometrium in Human Parturition. JCI Insight 2022, 7, e153921. [Google Scholar] [CrossRef]
- Chen, D.; Wang, W.; Wu, L.; Liang, L.; Wang, S.; Cheng, Y.; Zhang, T.; Chai, C.; Luo, Q.; Sun, C.; et al. Single-Cell Atlas of Peripheral Blood Mononuclear Cells from Pregnant Women. Clin. Transl. Med. 2022, 12, e821. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Wang, Y.-L.; Shi, W.; Hu, W.; Zeng, Z.; Liu, J.; Li, L.; Cai, W.; Tang, D.; Dai, Y. Multiplexed Analysis of Gene Expression and Chromatin Accessibility of Human Umbilical Cord Blood Using ScRNA-Seq and ScATAC-Seq. Mol. Immunol. 2022, 152, 207–214. [Google Scholar] [CrossRef]
- Shi, X.; Ma, W.; Duan, S.; Shi, Q.; Wu, S.; Hao, S.; Dong, G.; Li, J.; Song, Y.; Liu, C.; et al. Single-Cell Transcriptional Diversity of Neonatal Umbilical Cord Blood Immune Cells Reveals Neonatal Immune Tolerance. Biochem. Biophys. Res. Commun. 2022, 608, 14–22. [Google Scholar] [CrossRef]
- Yang, Y.; Guo, F.; Peng, Y.; Chen, R.; Zhou, W.; Wang, H.; OuYang, J.; Yu, B.; Xu, Z. Transcriptomic Profiling of Human Placenta in Gestational Diabetes Mellitus at the Single-Cell Level. Front. Endocrinol. 2021, 12, 679582. [Google Scholar] [CrossRef] [PubMed]
- Tsang, J.C.H.; Vong, J.S.L.; Ji, L.; Poon, L.C.Y.; Jiang, P.; Lui, K.O.; Ni, Y.-B.; To, K.F.; Cheng, Y.K.Y.; Chiu, R.W.K.; et al. Integrative Single-Cell and Cell-Free Plasma RNA Transcriptomics Elucidates Placental Cellular Dynamics. Proc. Natl. Acad. Sci. USA 2017, 114, E7786–E7795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.; Bian, Q.; Chen, Y.; Wang, X.; Yu, S.; Liu, S.; Ji, P.; Li, L.; Shrestha, M.; Dong, S.; et al. Dissecting Human Trophoblast Cell Transcriptional Heterogeneity in Preeclampsia Using Single-cell RNA Sequencing. Mol. Genet. Genom. Med. 2021, 9, e1730. [Google Scholar] [CrossRef]
- Zhou, W.; Wang, H.; Yang, Y.; Guo, F.; Yu, B.; Su, Z. Trophoblast Cell Subtypes and Dysfunction in the Placenta of Individuals with Preeclampsia Revealed by Single-Cell RNA Sequencing. Mol. Cells 2022, 45, 317–328. [Google Scholar] [CrossRef]
- Guo, C.; Cai, P.; Jin, L.; Sha, Q.; Yu, Q.; Zhang, W.; Jiang, C.; Liu, Q.; Zong, D.; Li, K.; et al. Single-Cell Profiling of the Human Decidual Immune Microenvironment in Patients with Recurrent Pregnancy Loss. Cell. Discov. 2021, 7, 1. [Google Scholar] [CrossRef]
- Wang, F.; Jia, W.; Fan, M.; Shao, X.; Li, Z.; Liu, Y.; Ma, Y.; Li, Y.-X.; Li, R.; Tu, Q.; et al. Single-Cell Immune Landscape of Human Recurrent Miscarriage. Genom. Proteom. Bioinform. 2021, 19, 208–222. [Google Scholar] [CrossRef]
- Du, L.; Deng, W.; Zeng, S.; Xu, P.; Huang, L.; Liang, Y.; Wang, Y.; Xu, H.; Tang, J.; Bi, S.; et al. Single-cell Transcriptome Analysis Reveals Defective Decidua Stromal Niche Attributes to Recurrent Spontaneous Abortion. Cell. Prolif. 2021, 54, e13125. [Google Scholar] [CrossRef] [PubMed]
- Pique-Regi, R.; Romero, R.; Tarca, A.L.; Sendler, E.D.; Xu, Y.; Garcia-Flores, V.; Leng, Y.; Luca, F.; Hassan, S.S.; Gomez-Lopez, N. Single Cell Transcriptional Signatures of the Human Placenta in Term and Preterm Parturition. eLife 2019, 8, e52004. [Google Scholar] [CrossRef]
- Liu, Q.; Li, Y.; Feng, Y.; Liu, C.; Ma, J.; Li, Y.; Xiang, H.; Ji, Y.; Cao, Y.; Tong, X.; et al. Single-Cell Analysis of Differences in Transcriptomic Profiles of Oocytes and Cumulus Cells at GV, MI, MII Stages from PCOS Patients. Sci. Rep. 2016, 6, 39638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, L.; Liu, B.; Chen, X.; Liu, Q.; Li, W.; Lv, B.; Xu, X.; Wang, L.; Zeng, Q.; Xue, J.; et al. Single-Cell Transcriptomic Analysis Reveals Mitochondrial Dynamics in Oocytes of Patients With Polycystic Ovary Syndrome. Front. Genet. 2020, 11, 396. [Google Scholar] [CrossRef]
- Ashary, N.; Bhide, A.; Chakraborty, P.; Colaco, S.; Mishra, A.; Chhabria, K.; Jolly, M.K.; Modi, D. Single-Cell RNA-Seq Identifies Cell Subsets in Human Placenta That Highly Expresses Factors Driving Pathogenesis of SARS-CoV-2. Front. Cell. Dev. Biol. 2020, 8, 783. [Google Scholar] [CrossRef]
- Zheng, Y.; Pan, J.; Xia, C.; Chen, H.; Zhou, H.; Ju, W.; Wegiel, J.; Myatt, L.; Roberts, J.M.; Guo, X.; et al. Characterization of Placental and Decidual Cell Development in Early Pregnancy Loss by Single-Cell RNA Sequencing. Cell. Biosci. 2022, 12, 168. [Google Scholar] [CrossRef] [PubMed]
- Caruso, M.; Evangelista, M.; Parolini, O. Human Term Placental Cells: Phenotype, Properties and New Avenues in Regenerative Medicine. Int. J. Mol. Cell. Med. 2012, 1, 64–74. [Google Scholar] [PubMed]
- Hammer, A. Immunological Regulation of Trophoblast Invasion. J. Reprod. Immunol. 2011, 90, 21–28. [Google Scholar] [CrossRef]
- Yang, F.; Zheng, Q.; Jin, L. Dynamic Function and Composition Changes of Immune Cells During Normal and Pathological Pregnancy at the Maternal-Fetal Interface. Front. Immunol. 2019, 10, 2317. [Google Scholar] [CrossRef] [Green Version]
- Balasundaram, P.; Farhana, A. Immunology at the Maternal-Fetal Interface. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Li, L.; Dong, J.; Yan, L.; Yong, J.; Liu, X.; Hu, Y.; Fan, X.; Wu, X.; Guo, H.; Wang, X.; et al. Single-Cell RNA-Seq Analysis Maps Development of Human Germline Cells and Gonadal Niche Interactions. Cell. Stem Cell. 2017, 20, 858–873.e4. [Google Scholar] [CrossRef] [Green Version]
- Rizvi, A.H.; Camara, P.G.; Kandror, E.K.; Roberts, T.J.; Schieren, I.; Maniatis, T.; Rabadan, R. Single-Cell Topological RNA-Seq Analysis Reveals Insights into Cellular Differentiation and Development. Nat. Biotechnol. 2017, 35, 551–560. [Google Scholar] [CrossRef] [PubMed]
- Jaitin, D.A.; Kenigsberg, E.; Keren-Shaul, H.; Elefant, N.; Paul, F.; Zaretsky, I.; Mildner, A.; Cohen, N.; Jung, S.; Tanay, A.; et al. Massively Parallel Single Cell RNA-Seq for Marker-Free Decomposition of Tissues into Cell Types. Science 2014, 343, 776–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segerstolpe, Å.; Palasantza, A.; Eliasson, P.; Andersson, E.-M.; Andréasson, A.-C.; Sun, X.; Picelli, S.; Sabirsh, A.; Clausen, M.; Bjursell, M.K.; et al. Single-Cell Transcriptome Profiling of Human Pancreatic Islets in Health and Type 2 Diabetes. Cell. Metab. 2016, 24, 593–607. [Google Scholar] [CrossRef] [Green Version]
- Grün, D.; Lyubimova, A.; Kester, L.; Wiebrands, K.; Basak, O.; Sasaki, N.; Clevers, H.; van Oudenaarden, A. Single-Cell Messenger RNA Sequencing Reveals Rare Intestinal Cell Types. Nature 2015, 525, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Guariguata, L.; Linnenkamp, U.; Beagley, J.; Whiting, D.R.; Cho, N.H. Global Estimates of the Prevalence of Hyperglycaemia in Pregnancy. Diabetes Res. Clin. Pract. 2014, 103, 176–185. [Google Scholar] [CrossRef]
- Plows, J.F.; Stanley, J.L.; Baker, P.N.; Reynolds, C.M.; Vickers, M.H. The Pathophysiology of Gestational Diabetes Mellitus. Int. J. Mol. Sci. 2018, 19, 3342. [Google Scholar] [CrossRef] [Green Version]
- Lu, W.; Hu, C. Molecular Biomarkers for Gestational Diabetes Mellitus and Postpartum Diabetes. Chin. Med. J. 2022, 135, 1940–1951. [Google Scholar] [CrossRef]
- Monod, C.; Kotzaeridi, G.; Linder, T.; Eppel, D.; Rosicky, I.; Filippi, V.; Tura, A.; Hösli, I.; Göbl, C.S. Prevalence of Gestational Diabetes Mellitus in Women with a Family History of Type 2 Diabetes in First- and Second-Degree Relatives. Acta Diabetol. 2023, 60, 345–351. [Google Scholar] [CrossRef]
- Robitaille, J.; Grant, A.M. The Genetics of Gestational Diabetes Mellitus: Evidence for Relationship with Type 2 Diabetes Mellitus. Genet. Med. 2008, 10, 240–250. [Google Scholar] [CrossRef] [Green Version]
- Leoni, M.; Padilla, N.; Fabbri, A.; Della-Morte, D.; Ricordi, C.; Infante, M.; Leoni, M.; Padilla, N.; Fabbri, A.; Della-Morte, D.; et al. Mechanisms of Insulin Resistance during Pregnancy. In Evolving Concepts in Insulin Resistance; IntechOpen: London, UK, 2022; ISBN 978-1-80355-502-7. [Google Scholar]
- Lawlor, N.; George, J.; Bolisetty, M.; Kursawe, R.; Sun, L.; Sivakamasundari, V.; Kycia, I.; Robson, P.; Stitzel, M.L. Single-Cell Transcriptomes Identify Human Islet Cell Signatures and Reveal Cell-Type-Specific Expression Changes in Type 2 Diabetes. Genome Res. 2017, 27, 208–222. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Pan, X.; Cai, Y.-D. Identification of Type 2 Diabetes Biomarkers from Mixed Single-Cell Sequencing Data With Feature Selection Methods. Front. Bioeng. Biotechnol. 2022, 10, 890901. [Google Scholar] [CrossRef]
- Phipps, E.A.; Thadhani, R.; Benzing, T.; Karumanchi, S.A. Pre-Eclampsia: Pathogenesis, Novel Diagnostics and Therapies. Nat. Rev. Nephrol. 2019, 15, 275–289. [Google Scholar] [CrossRef]
- Suman, V.; Luther, E.E. Preterm Labor. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Lucaroni, F.; Morciano, L.; Rizzo, G.; D’ Antonio, F.; Buonuomo, E.; Palombi, L.; Arduini, D. Biomarkers for Predicting Spontaneous Preterm Birth: An Umbrella Systematic Review. J. Matern. Fetal Neonatal Med. 2018, 31, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Definitions of Infertility and Recurrent Pregnancy Loss: A Committee Opinion. Fertil. Steril. 2013, 99, 63. [CrossRef] [PubMed]
- El Hachem, H.; Crepaux, V.; May-Panloup, P.; Descamps, P.; Legendre, G.; Bouet, P.-E. Recurrent Pregnancy Loss: Current Perspectives. Int. J. Women’s Health 2017, 9, 331–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allison, J.L.; Schust, D.J. Recurrent First Trimester Pregnancy Loss: Revised Definitions and Novel Causes. Curr. Opin. Endocrinol. Diabetes Obes. 2009, 16, 446–450. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Gimenez, C.; Alijotas-Reig, J. Recurrent Miscarriage: Causes, Evaluation and Management. Postgrad. Med. J. 2015, 91, 151–162. [Google Scholar] [CrossRef]
- Li, J.; Wang, L.; Ding, J.; Cheng, Y.; Diao, L.; Li, L.; Zhang, Y.; Yin, T. Multiomics Studies Investigating Recurrent Pregnancy Loss: An Effective Tool for Mechanism Exploration. Front. Immunol. 2022, 13, 826198. [Google Scholar] [CrossRef]
- Escobar-Morreale, H.F. Polycystic Ovary Syndrome: Definition, Aetiology, Diagnosis and Treatment. Nat. Rev. Endocrinol. 2018, 14, 270–284. [Google Scholar] [CrossRef]
- Kamalanathan, S.; Sahoo, J.P.; Sathyapalan, T. Pregnancy in Polycystic Ovary Syndrome. Indian J. Endocrinol. Metab. 2013, 17, 37–43. [Google Scholar] [CrossRef] [PubMed]
- March, W.A.; Moore, V.M.; Willson, K.J.; Phillips, D.I.W.; Norman, R.J.; Davies, M.J. The Prevalence of Polycystic Ovary Syndrome in a Community Sample Assessed under Contrasting Diagnostic Criteria. Hum. Reprod. 2010, 25, 544–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babayev, E.; Seli, E. Oocyte Mitochondrial Function and Reproduction. Curr. Opin. Obstet. Gynecol. 2015, 27, 175–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
| Year Ref. | Sample Type | Research Group | Gestational Age | Number of Cells | Method | Main Bioinformatic Tools * | Main Findings |
|---|---|---|---|---|---|---|---|
| Normal pregnancy | |||||||
| 2018 [60] | Placenta | Normal pregnancy (n = 8) | 8 and 24 weeks | 1567 | MACS, smart-seq2 | TopHat, HTSeq, Seurat, Monocle2, KEGG | Fourteen subtypes of placental cells: three CTBs1 subtypes, STBs2 subtype, EVTs3 subtypes, two macrophages’ cells subtypes, two mesenchymal stromal cells subtypes, one blood cell subtype in the villi, and two EVTs3 subtypes in the decidua. |
| 2018 [61] | Villi and decidua | Villi (n = 8), decidua (n = 6), | 6–11 weeks, elective termination | 21,095 | Cell counter, Drop-seq, 10X Genomics | STAR, featureCounts, Seurat, DAVID | Transcriptome definition of 20 cell populations; the relative proportions of each cell type in villi and decidual samples; an interactome map between the most abundantly expressed ligands and receptors in villi and decidua cells; the new subtypes of the FB4-like cells. |
| 2018 [21] | Placenta, decidua cells, maternal PBMC7 | Decidua (n = 11), placenta (n = 5), PBMC (n = 6) | 6–14 weeks | >70,000 | FACS, 10X Genomics, smartSeq2 | Cell Ranger, HISAT2, HTSeq, Seurat, Monocle2, Cytoscape, CellPhoneDB | Molecular and cellular map of the human decidual-placental interface; characterization of three DSC5 cell subtypes (dS1, dS2, and dS3) and three dNK6 subtypes; differentiation trajectory from CTB1 to EVT3; repository of ligand-receptor complexes to predict interactions between different cells of the maternal-fetal interface (CellPhoneDB). |
| 2022 [62] | Placenta | After delivery (n = 8), full-term | 38–40 weeks | 11,438 | MACS, 10X Genomics | Cell Ranger, Seurat, Monocle2, scanpy, clusterProfiler, CellPhoneDB | The maternal–fetal interface cellular map of full-term placenta; a subpopulation of TPLCs8 with high expression of HMMR; downregulation of PRDM6 may lead to an abnormal endovascular EVTs3 differentiation process in preeclampsia. |
| 2023 [63] | Villi | Normal pregnancy (n = 11), | 6–16 weeks, elective termination | 52,179 | HTBS9, 10X Genomics | Cell Ranger, Seurat, Monocle 2, CellPhoneDB | Three new populations of progenitor cells: endothelial progenitors, STB2 progenitors, and EVT3 progenitors; 8–9 gestational weeks were determined as a critical time point for altering gene expression profiles in placental cells. |
| 2022 [64] | Myometrium | Term in spontaneous labor (n = 11), term not in labor (n = 13), | ≥37 weeks, caesarean section | 53,194 | Cellometer Auto 2000; 10X Genomics | Cell Ranger, kallisto, bus tools, STAR, SingleR, Seurat, DESeq2, clusterProfiler, SPSS | A single-cell atlas of the human myometrium; cell–cell communications that are modulated during the physiologic process of spontaneous labor at term; ERRFI1, a specifically differentially expressed gene in maternally circulating monocytes; nonimmune and immune cells participate in a plethora of biological pathways associated with the contractile and inflammatory processes of spontaneous labor at term. |
| 2022 [65] | PBMC | Normal pregnancy (n = 131), non-pregnancy (n = 5) | 6–40 weeks | 198,356 | HTBS9, MGI DNBelab, TF Scientific | PISA, Seurat, clusterProfiler, CellChat, MAGIC algorithm, SHAP | A single-cell atlas of PBMCs7 in pregnant women spanning the entire gestation period; cell-type-specific model to predict gestational age in normal pregnancy; interferon-stimulated gene upregulation. |
| 2022 [66] | UCB10 cells | Normal (n = 4) | 31–37 weeks, after delivery | 3866 | Countess II, 10X Genomics | Cell Ranger | New cell types (erythroid cell, T cell, B cell, erythroid precursor cells, NK cell, and endothelial progenitor cell), new subpopulations (six different clusters of erythroid cells) in UCB10; the differentially expressed genes and chromatin accessibility in each cell between different gestational weeks. |
| 2022 [67] | UCB10 cells | Normal (n = 3) | After delivery | 57,467 | DNBelab C | STAR, PISA, Seurat, UMAP, bap2, clusterProfiler | Differential gene expression regulation between neonatal and adult T and B cells; the global molecular features of transcription and chromatin accessibility in neonatal UCB10 nucleated cells and adult PBMCs7. |
| Gestational diabetes mellitus | |||||||
| 2021 [68] | Placenta | Gestational diabetes mellitus (n = 20), normal (n = 20) | Full-term, caesarean section | 27,220 | HTBS9, 10X Genomics | Cell Ranger, Seurat, SingleR, Monocle2, SCENIC, CellPhoneDB, Velocyto, GSVA | The comprehensive cell atlas for the gestational diabetes mellitus placenta; characterization of nine cell types in the human placenta; a significant increase of NK and cytotoxic T cells, enhancement of M2 macrophages, and decrease of inflammatory response cells in the gestational diabetes mellitus placenta; ligand-receptor interactions in the maternal and fetal microenvironment, as well as new marker genes, including SLC1A2, SLC1A6, ADRB1. |
| Preeclampsia | |||||||
| 2017 [69] | Placenta | Early-onset preeclampsia (n = 4), normal (n = 6) | 28–32 weeks, healthy 38 weeks, cesarean section | >24,000 | 10X Genomics | Cell Ranger, STAR, Rtsne | A large-scale single-cell transcriptomic atlas of the normal and early preeclamptic placentas; the differentiation relationships between the CTBs1, STBs2, and EVTs3 were re-confirmed; a significant increase of variability and levels expression of cell death-related genes in early preeclamptic EVTs3; plasma cell-free RNAs may be useful as markers of placenta cellular composition and preeclampsia. |
| 2021 [70] | Placenta | Preeclampsia (n = 3), normal (n = 3) | 34–38 weeks, cesarean section | 11,518 | Singlerone GEXSCOPE | Ensembl, fastp, featureCounts, Seurat, clusterProfiler, Monocle 2, DDRTree | Differences in transcriptional profiles of STBs2, EVTs3, and VCTs11 between preeclampsia and healthy patients; VCTs11 and EVTs3 show immune response in preeclampsia; signaling pathways in STBs2 upregulated in the preeclampsia; three new VCTs11 subtypes; a significant increase of VCT-2 cells in the preeclampsia placenta. |
| 2022 [71] | Placenta | Early-onset preeclampsia (n = 2), healthy (n = 2) | 32–40 weeks, cesarean section | 29,008 | HTBS9, 10X Genomics | Cell Ranger, Seurat, SCENIC, scFunctions, GSEA, Cytoscape | Differences in transcriptional profiles of STBs2, EVTs3, and VCTs11 between preeclampsia and healthy patients; two new transcriptional factors, CEBPB and GTF2B, involved in EVTs3 dysfunction in preeclampsia. |
| Recurrent pregnancy loss | |||||||
| 2021 [72] | Decidua | Recurrent pregnancy loss (n = 9), healthy (n = 15) | 7–9 weeks | 18,646 | FACS; 10X Genomics | Cell Ranger, Seurat, SAVER, velocyto, CellPhoneDB, Cytoscape | Changes in the number of dNK6 cells and macrophages function between recurrent pregnancy loss and normal pregnancy; a decrease of macrophage populations in recurrent pregnancy loss; a significant decrease of dNK6 subset with growth-supporting activity and an increase of pro-inflammatory dNK6 subset that produces cytokines in recurrent pregnancy loss; ligand/receptor level hypothesis about the likely causes underlying pregnancy failure. |
| 2021 [73] | PBMC7 decidua | Recurrent miscarriage (n = 14), normal (n = 10) | 6–8 weeks, therapeutic termination | 56,758 | 10X Genomics | Cell Ranger, Seurat | A comprehensive cellular and molecular atlas of decidual and peripheral leukocytes in early pregnancy; an increase of dNK3 subset with cytotoxic and immune-active; the unique accumulation of the dNK4 subset with pro-inflammatory properties in the recurrent miscarriages; increased expression of inflammation-related genes in dNK6 cells from recurrent miscarriages; cytotoxic properties of T cells, NK-cells, and mucosal-associated invariant T cells in peripheral blood. |
| 2021 [74] | Decidua | Recurrent spontaneous abortion (n = 6), normal (n = 5) | 5–8 weeks | 66,078 | 10X Genomics | Cell Ranger, STAR, Seurat, Monocle 2, CellPhoneDB, CellChat, SCENIC | Characterization of the five clusters of DSCs5; changes in the number of decidualized stromal cells in recurrent spontaneous abortion; cell composition and communications in normal and recurrent spontaneous abortion decidua at early pregnancy; the aberrant decidualization and obstructed communication between stromal cells accompanying recurrent spontaneous abortion. |
| Preterm labor | |||||||
| 2019 [75] | Placenta | Preterm labor (n = 3), term no labor (n = 3), term in labor (n = 3), | Preterm (33–35 weeks), term labor (38–40 weeks) | 79,906 | Cellometer Auto 2000; 10X Genomics | Cell Ranger, STAR, Seurat, xCell, DESeq2 | Two cell types: lymphatic endothelial decidual cells in the chorioamniotic membranes and non-proliferative interstitial cytotrophoblasts in the placental villi; a significant increase of NFKB1 gene in macrophages from women with preterm labor. |
| Conditions associated with an increased risk of pregnancy complications | |||||||
| 2016 [76] | Cumulus- oocyte complex | Polycystic ovary syndrome (n = 9), healthy (n = 7) | - | 28 cumulus cells | Smart-seq2 | Read alignment and quantification methods not disclosed; DAVID | Differentially expressed genes, including PPP2R1A, PDGFRA, EGFR, PTGS, CAV1, INHBB, etc., detected as potential causes of PCOS oocytes and CCs disorder at early stages; restoration of their normal expression level via assisted reproductive techniques, which can be an effective treatment for subfertile patients with PCOS. |
| 2020 [77] | Cumulus- oocyte complex | Polycystic ovary syndrome (n = 9), healthy (n = 7) | - | 28 cumulus cells | Smart-seq2 | Read alignment and quantification methods not disclosed; DAVID, DESeq2, WGCNA, Cytoscape, GSEA, clusterProfiler, | Downregulation of CYP26A1, MTRNR2L1, and ELOA genes, upregulation of FAM53A, PPP1R35, and BLM in PCOS oocytes; potential premature activation of mitochondrial function in PCOS oocytes. |
| 2020 [78] | Placenta | Healthy patients (n = 8) | 8–24 weeks | 1567 | MACS, smart-seq2 | TopHat, HTSeq, Seurat, Monocle2, ARACNe-AP, KEGG | ACE2 expression in EVTs3 of the first and second trimester placenta; BSG/CD147, the alternate receptor for SARS-CoV-2, expressed by almost all the placental cells; an abundant expression of DPP4 (MERS-CoV receptor) and ANPEP (CoV-229E receptor) in the cells of the placenta; co-expression of BSG/CD147 with ACE2 in STBs2 and EVTs3; an increased incidence of preterm delivery in women with COVID-19 was assumed. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naydenov, D.D.; Vashukova, E.S.; Barbitoff, Y.A.; Nasykhova, Y.A.; Glotov, A.S. Current Status and Prospects of the Single-Cell Sequencing Technologies for Revealing the Pathogenesis of Pregnancy-Associated Disorders. Genes 2023, 14, 756. https://doi.org/10.3390/genes14030756
Naydenov DD, Vashukova ES, Barbitoff YA, Nasykhova YA, Glotov AS. Current Status and Prospects of the Single-Cell Sequencing Technologies for Revealing the Pathogenesis of Pregnancy-Associated Disorders. Genes. 2023; 14(3):756. https://doi.org/10.3390/genes14030756
Chicago/Turabian StyleNaydenov, Dmitry D., Elena S. Vashukova, Yury A. Barbitoff, Yulia A. Nasykhova, and Andrey S. Glotov. 2023. "Current Status and Prospects of the Single-Cell Sequencing Technologies for Revealing the Pathogenesis of Pregnancy-Associated Disorders" Genes 14, no. 3: 756. https://doi.org/10.3390/genes14030756