
Pathophysiological Investigation of Skeletal Deformities of Musculocontractural Ehlers–Danlos Syndrome Using Induced Pluripotent Stem Cells

Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Source
2.2. Generation of iPSC Lines
2.3. Sanger Sequencing
2.4. Teratoma Formation
2.5. Osteogenic Differentiation of iPSCs
2.6. Immunocytochemistry
2.7. Quantitative Reverse Transcription PCR (qRT-PCR)
2.8. Alizarin Red Staining
2.9. Quantification of Calcium Deposition
2.10. Statistical Analysis
3. Results
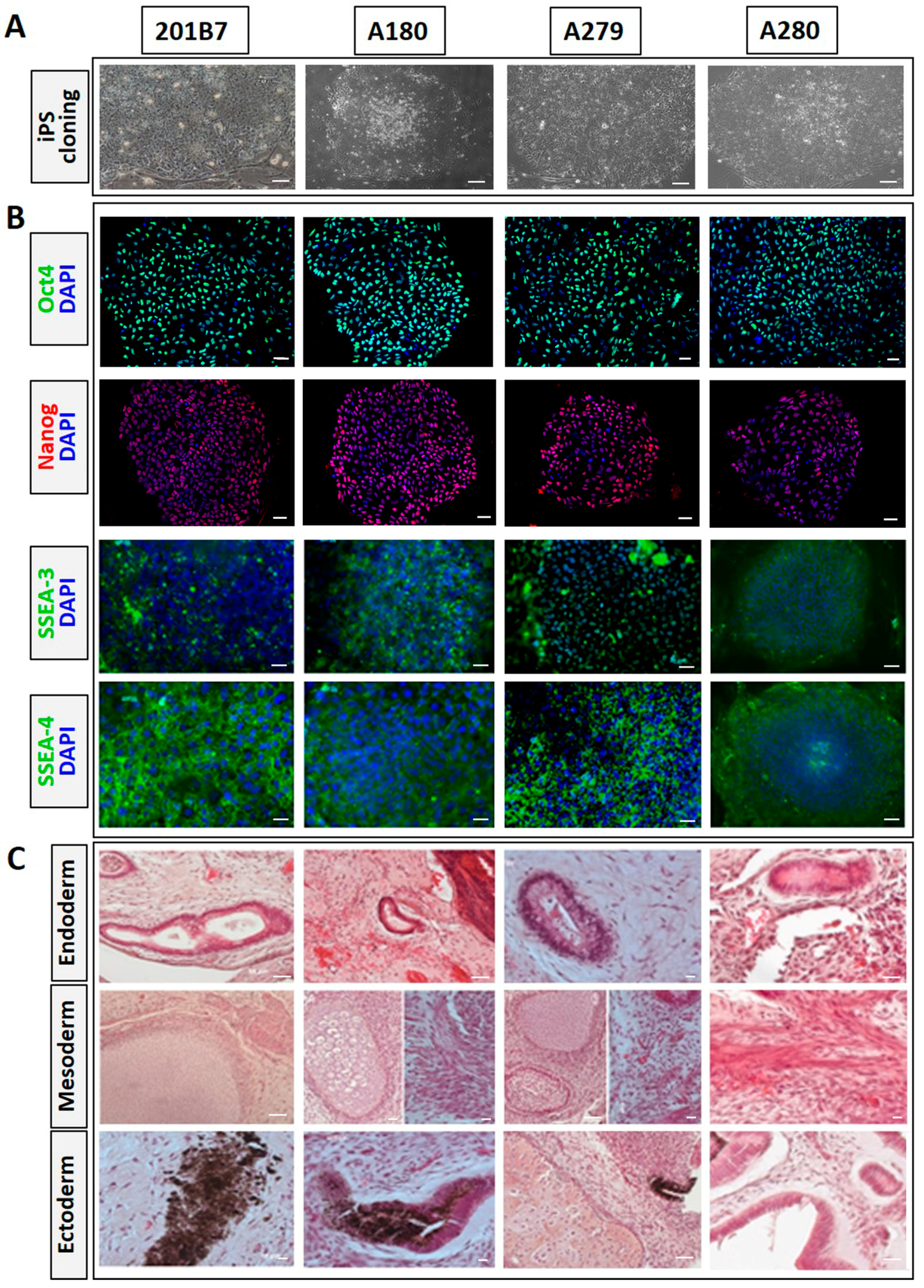
3.1. Generation of iPSCs from Skin Fibroblasts of mcEDS-CHST14 Patients
3.2. Identification of CHST14 Gene Mutation
3.3. Sequential Differentiation of mcEDS-CHST14 iPSCs into the Osteogenic Lineage
3.4. Impaired Osteogenesis of iPSCs Derived from mcEDS-CHST14 Patients
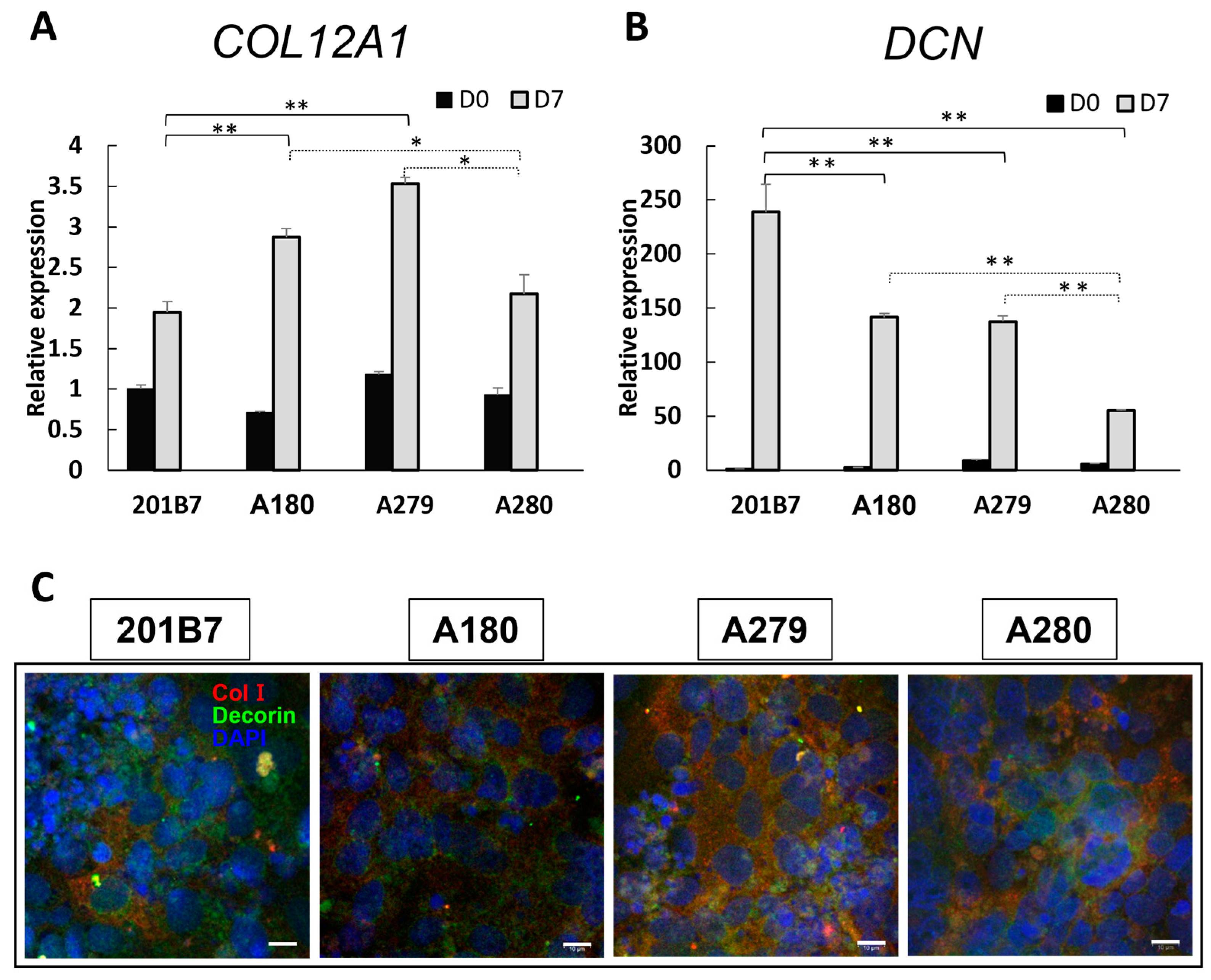
3.5. Expression of Collagen and Decorin
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Malfait, F.; Castori, M.; Francomano, C.A.; Giunta, C.; Kosho, T.; Byers, P.H. The Ehlers-Danlos syndromes. Nat. Rev. Dis. Prim. 2020, 6, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Malfait, F.; Francomano, C.; Byers, P.; Belmont, J.; Berglund, B.; Black, J.; Bloom, L.; Bowen, J.M.; Brady, A.F.; Burrows, N.P.; et al. The 2017 international classification of the Ehlers-Danlos syndromes. Am. J. Med. Genet. Part C Semin. Med. Genet. 2017, 175, 8–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brady, A.F.; Demirdas, S.; Fournel-Gigleux, S.; Ghali, N.; Giunta, C.; Kapferer-Seebacher, I.; Kosho, T.; Mendoza-Londono, R.; Pope, M.F.; Rohrbach, M.; et al. The Ehlers-Danlos syndromes, rare types. Am. J. Med. Genet. Part C Semin. Med. Genet. 2017, 175, 70–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minatogawa, M.; Unzaki, A.; Morisaki, H.; Syx, D.; Sonoda, T.; Janecke, A.R.; Slavotinek, A.; Voermans, N.C.; Lacassie, Y.; Mendoza-Londono, R.; et al. Clinical and molecular features of 66 patients with musculocontractural Ehlers-Danlos syndrome caused by pathogenic variants in CHST14 (mcEDS-CHST14). J. Med. Genet. 2022, 59, 865–877. [Google Scholar] [CrossRef] [PubMed]
- Minatogawa, M.; Hirose, T.; Mizumoto, S.; Yamaguchi, T.; Nagae, C.; Taki, M.; Yamada, S.; Watanabe, T.; Kosho, T. Clinical and pathophysiological delineation of musculo-contractural Ehlers-Danlos syndrome caused by dermatan sulfate epimerase deficiency (mcEDS-DSE): A detailed and comprehensive glycobiological and pathological investigation in a novel patient. Hum. Mutat. 2022, 43, 1829–1836. [Google Scholar] [CrossRef]
- Evers, M.R.; Xia, G.; Kang, H.G.; Schachner, M.; Baeziger, J.U. Molecular cloning and characterization of a dermatan-specific N-acetylgalactosamine 4-O-sulfotransferase. J. Biol. Chem. 2001, 276, 36344–36353. [Google Scholar] [CrossRef] [Green Version]
- Mikami, T.; Mizumoto, S.; Kago, N.; Kitagawa, H.; Sugahara, K. Specificities of three distinct human chondroitin/dermatan N-acetylgalactosamine 4-O-sulfotransferases demonstrated using partially desulfated dermatan sulfate as an acceptor: Implication of differential roles in dermatan sulfate biosynthesis. J. Biol. Chem. 2003, 278, 36115–36127. [Google Scholar] [CrossRef] [Green Version]
- Trowbridge, J.M.; Gallo, R.L. Dermatan sulfate: New functions from an old glycosaminoglycan. Glycobiology 2002, 12, 117–125. [Google Scholar] [CrossRef]
- Zhang, L.; Müller, T.; Baenziger, J.U.; Janecke, A.R. Congenital disorders of glycosylation with emphasis on loss of dermatan-4-sulfotransferase? Prog. Mol. Biol. Transl. Sci. 2010, 93, 289–307. [Google Scholar] [CrossRef]
- Shuaa, B.; Frank, R. Bone Disease in Patients with Ehlers-Danlos Syndromes. Curr. Osteoporos. Rep. 2020, 18, 95–102. [Google Scholar] [CrossRef]
- Uehara, M.; Kosho, T.; Yamamoto, N.; Takahashi, H.E.; Shimakura, T.; Nakayama, J.; Kato, H.; Takahashi, J. Spinal manifestations in 12 patients with musculocontractural Ehlers-Danlos syndrome caused by CHST14/D4ST1 deficiency (mcEDS-CHST14). Am. J. Med. Genet. A 2018, 176, 2331–2341. [Google Scholar] [CrossRef] [PubMed]
- Robey, P.G.; Bianco, P.; Termine, J.D. The cellular biology and molecular biochemistry of bone formation. In Disorders of Bone Mineral Metabolism; Coe, F.L., Favus, M.J., Eds.; Raven Press: New York, NY, USA, 1992; pp. 241–263. [Google Scholar]
- Nitahara-Kasahara, Y.; Mizumoto, S.; Inoue, Y.U.; Saka, S.; Posadas-Herrera, G.; Nakamura-Takahashi, A.; Takahashi, Y.; Hashimoto, A.; Konishi, K.; Miyata, S.; et al. A new mouse model of Ehlers-Danlos syndrome generated using CRISPR/Cas9-mediated genomic editing. Dis. Model. Mech. 2021, 14, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Yoshizawa, T.; Mizumoto, S.; Takahashi, Y.; Shimada, S.; Sugahara, K.; Nakayama, J.; Takeda, S.; Nomura, Y.; Nitahara-Kasahara, Y.; Okada, T.; et al. Vascular abnormalities in the placenta of Chst14-/- fetuses: Implications in the pathophysiology of perinatal lethality of the murine model and vascular lesions in human CHST14/D4ST1 deficiency. Glycobiology 2018, 28, 80–89. [Google Scholar] [CrossRef] [Green Version]
- Ohnishi, K.; Semi, K.; Yamamoto, T.; Shimizu, M.; Tanaka, A.; Mitsunaga, K.; Okita, K.; Osafune, K.; Arioka, Y.; Maeda, T.; et al. Premature termination of reprogramming in vivo leads to cancer development through altered epigenetic regulation. Cell 2014, 156, 663–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, H.; Lensch, M.W.; Cahan, P.; Daley, G.Q. Investigating monogenic and complex diseases with pluripotent stem cells. Nat. Rev. Genet. 2011, 12, 266–275. [Google Scholar] [CrossRef]
- Duan, X.; Tu, Q.; Zhang, J.; Ye, J.; Sommer, C.; Mostoslavsky, G.; Kaplan, D.; Yang, P.; Chen, J. Application of induced pluripotent stem (iPS) cells in periodontal tissue regeneration. J. Cell. Physiol. 2011, 226, 150–157. [Google Scholar] [CrossRef] [Green Version]
- Rowe, R.G.; Daley, G.Q. Induced pluripotent stem cells in disease modelling and drug discovery. Nat. Rev. Genet. 2019, 20, 377–388. [Google Scholar] [CrossRef]
- Kosho, T.; Takahashi, J.; Ohashi, H.; Nishimura, G.; Kato, H.; Fukushima, Y. Ehlers-Danlos syndrome type VIB with characteristic facies, decreased curvatures of the spinal column, and joint contractures in two unrelated girls. Am. J. Med. Genet. A 2005, 138A, 282–287. [Google Scholar] [CrossRef]
- Kosho, T.; Miyake, N.; Hatamochi, A.; Takahashi, J.; Kato, H.; Miyahara, T.; Igawa, Y.; Yasui, H.; Ishida, T.; Ono, K.; et al. A new Ehlers-Danlos syndrome with craniofacial characteristics, multiple congenital contractures, progressive joint and skin laxity, and multisystem fragility-related manifestations. Am. J. Med. Genet. A 2010, 152A, 1333–1346. [Google Scholar] [CrossRef]
- Isobe, F.; Hayashi, M.; Kobayashi, R.; Nakamura, M.; Kosho, T.; Takahashi, J. Clinical presentation and characteristics of the upper extremity in patients with musculocontractural Ehlers-Danlos Syndrome. Genes 2022, 13, 1978. [Google Scholar] [CrossRef]
- Shimizu, K.; Okamoto, N.; Miyake, N.; Taira, K.; Sato, Y.; Matsuda, K.; Akimaru, N.; Ohashi, H.; Wakui, K.; Fukushima, Y.; et al. Delineation of dermatan 4-O-sulfotransferase 1 deficient Ehlers-Danlos syndrome: Observation of two additional patients and comprehensive review of 20 reported patients. Am. J. Med. Genet. Part A 2011, 155A, 1949–1958. [Google Scholar] [CrossRef] [PubMed]
- Mizumoto, S.; Kosho, T.; Honda, T.; Hatamochi, A.; Sugahara, K.; Matsumoto, N.; Miyake, N.; Yamada, S. Biochemical characterization of an atypical variant flanking or affecting the initiation or start methionine codon in CHST14 in a patient with musculocontractural Ehlers−Danlos Syndrome. Front. Genet. 2023; submitted. [Google Scholar]
- Kawai, S.; Yoshitomi, H.; Sunaga, J.; Alev, C.; Nagata, S.; Nishio, M.; Hada, M.; Koyama, Y.; Matsuda, S. In vitro bone-like nodules generated from patient-derived iPSCs recapitulate pathological bone phenotypes. Nat. Biomed. Eng. 2019, 3, 558–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takizawa, S.; Yoshie, S.; Yue, F.; Mogi, A.; Yokoyama, T.; Tomotsune, D.; Sasaki, K. FGF7 and cell density are required for final differentiation of pancreatic amylase-positive cells from human ES cells. Cell Tissue Res. 2013, 354, 751–759. [Google Scholar] [CrossRef]
- Shi, G.; Jin, Y. Role of Oct4 in maintaining and regaining stem cell pluripotency. Stem Cell Res. Ther. 2010, 1, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Komori, T. Regulation of bone development and extracellular matrix protein genes by RUNX2. Cell Tissue Res. 2010, 339, 189–195. [Google Scholar] [CrossRef] [Green Version]
- Zou, L.; Kidwai, F.K.; Kopher, R.A.; Motl, J.; Kellum, C.A.; Westendorf, J.J.; Kaufman, D.S. Use of RUNX2 expression to identify osteogenic progenitor cells derived from human embryonic stem cells. Stem Cell Rep. 2015, 4, 190–198. [Google Scholar] [CrossRef] [Green Version]
- Wei, J.; Karsenty, G. An overview of the metabolic functions of osteocalcin. Rev. Endocr. Metab. Disord. 2015, 16, 93–98. [Google Scholar] [CrossRef] [Green Version]
- Feng, J.; Clinkenbeard, E.L.; Yuan, B.; White, K.E.; Drezne, M.K. Osteocyte regulation of phosphate homeostasis and bone mineralization underlies the pathophysiology of the heritable disorders of rickets and osteomalacia. Bone 2013, 54, 213–221. [Google Scholar] [CrossRef] [Green Version]
- Hirose, T.; Takahashi, N.; Tangkawattana, P.; Minaguchi, J.; Mizumoto, S.; Yamada, S.; Miyake, N.; Hayashi, S.; Hatamochi, A.; Nakayama, J.; et al. Structural alteration of glycosaminoglycan side chains and spatial disorganization of collagen networks in the skin of patients with mcEDS-CHST14. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 623–631. [Google Scholar] [CrossRef]
- Carrin, S.V.; Garnero, P.; Delmas, P.D. The role of collagen in bone strength. Osteoporos. Int. 2006, 17, 319–336. [Google Scholar] [CrossRef] [PubMed]
- Tzaphlidou, M. Bone Architecture: Collagen Structure and Calcium/Phosphorus Maps. J. Biol. Phys. 2008, 34, 39–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forlino, A.; Cabral, W.A.; Barnes, A.M.; Marini, J.C. New Perspectives on Osteogenesis Imperfecta. Nat. Rev. Endocrinol. 2011, 14, 540–557. [Google Scholar] [CrossRef] [Green Version]
- Galicka, A.G. Mutations in type I collagen genes resulting in osteogenesis imperfecta in humans. Acta Biochim. Pol. 2002, 49, 433–441. [Google Scholar] [CrossRef] [Green Version]
- Hocking, A.M.; Shinomura, T.; McQuillan, D.J. Leucine-rich repeat glycoproteins of the extracellular matrix. Matrix Biol. 1998, 17, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Bianco, P.; Fisher, L.W.; Young, M.F.; Termine, J.D.; Robey, P.G. Expression and localization of the two small proteoglycans biglycan and decorin in developing human skeletal and non-skeletal tissues. J. Histochem. Cytochem. 1990, 38, 1549–1563. [Google Scholar] [CrossRef] [Green Version]
- Schuh, K.; Uldrijan, S.; Gambaryan, S.; Roethlein, N.; Neyses, L. Interaction of the plasma membrane Ca2+ pump 4b/CI with the Ca2+/calmodulin-dependent membrane-associated kinase CASK. J. Biol. Chem. 2003, 278, 9778–9783. [Google Scholar] [CrossRef] [Green Version]
- Fischer, J.W.; Kinsella, M.G.; Levkau, B.; Clowes, A.W.; Wight, T.N. Retroviral overexpression of decorin differentially affects the response of arterial smooth muscle cells to growth factors. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 777–784. [Google Scholar] [CrossRef] [Green Version]
- Kamiya, N.; Shigemasa, K.; Takagi, M. Gene expression and immunohistochemical localization of decorin and biglycan in association with early bone formation in the developing mandible. J. Oral Sci. 2001, 43, 179–188. [Google Scholar] [CrossRef]
- Kim, D.; Choi, J.; Han, K.M.; Lee, B.H.; Choi, J.H.; Yoo, H.W.; Han, Y.M. Impaired osteogenesis in Menkes disease-derived induced pluripotent stem cells. Stem Cell Res. Ther. 2015, 6, 161–166. [Google Scholar] [CrossRef] [Green Version]
- Hirose, T.; Mizumoto, S.; Hashimoto, A.; Takahashi, Y.; Yoshizawa, T.; Nitahara-Kasahara, Y.; Takahashi, N.; Nakayama, J.; Takehana, K.; Okada, T.; et al. Systematic investigation of the skin in Chst14-/- mice: A model for skin fragility in musculocontractural Ehlers-Danlos syndrome caused by CHST14 variants (mcEDS-CHST14). Glycobiology 2021, 31, 137–150. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, A.; Hirose, T.; Hashimoto, K.; Mizumoto, S.; Nitahara-Kasahara, Y.; Saka, S.; Yoshizawa, T.; Okada, T.; Yamada, S.; Kosho, T.; et al. Collagen network formation in in vitro models of musculocontractural Ehlers-Danlos syndrome. Genes 2023, 14, 308. [Google Scholar] [CrossRef] [PubMed]
- Syx, D.; Van Damme, T.; Symoens, S.; Maiburg, M.C.; van de Laar, I.; Morton, J.; Suri, M.; Del Campo, M.; Hausser, I.; Hermanns-Lê, T.; et al. Genetic heterogeneity and clinical variability in musculocontractural Ehlers-Danlos syndrome caused by impaired dermatan sulfate biosynthesis. Hum. Mutat. 2015, 36, 535–547. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient ID | A180 | A279 | A280 |
|---|---|---|---|
| Coding DNA change (NM_130468.4) | c.[842C>T];[878A>G] | c.[842C>T];[878A>G] | c.[2_10del];[2_10del] |
| Protein alteration (NP_569735.1) | p.[Pro281Leu];[Tyr293Cys] | p.[Pro281Leu];[Tyr293Cys] | p.[?];[?] |
| Age at skin biopsy | 18 y | 3 y | 12 y |
| Sex | Female | Male | Male |
| Ethnicity | Japanese | Japanese | Japanese |
| Craniofacial features | Typical | Typical | Typical |
| Congenital contractures | Adducted thumb (bil) Clubfeet (lt) | Adducted thumb (bil) Clubfeet (bil) | Adducted thumb (bil) |
| Spinal deformity | Kyphoscoliosis (severe, progressive) | Kyphoscoliosis (mild) | Kyphoscoliosis (severe, progressive) |
| Skin features | Typical | Typical | Typical |
| Large subcutaneous hematomas | Severe, recurrent | Severe, recurrent | Severe, recurrent |
| References | Kosho et al., 2005 [19] Kosho et al., 2010 [20] Uehara et al., 2018 [11] Minatogawa et al., 2022 [4] Isobe et al., 2022 [21] | Shimizu et al., 2011 [22] Uehara et al., 2018 [11] Isobe et al., 2022 [21] Minatogawa et al., 2022 [4] | Uehara et al., 2018 [11] Minatogawa et al., 2022 [4] Isobe et al., 2022 [21] Mizumoto et al., 2023 [23] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yue, F.; Era, T.; Yamaguchi, T.; Kosho, T. Pathophysiological Investigation of Skeletal Deformities of Musculocontractural Ehlers–Danlos Syndrome Using Induced Pluripotent Stem Cells. Genes 2023, 14, 730. https://doi.org/10.3390/genes14030730
Yue F, Era T, Yamaguchi T, Kosho T. Pathophysiological Investigation of Skeletal Deformities of Musculocontractural Ehlers–Danlos Syndrome Using Induced Pluripotent Stem Cells. Genes. 2023; 14(3):730. https://doi.org/10.3390/genes14030730
Chicago/Turabian StyleYue, Fengming, Takumi Era, Tomomi Yamaguchi, and Tomoki Kosho. 2023. "Pathophysiological Investigation of Skeletal Deformities of Musculocontractural Ehlers–Danlos Syndrome Using Induced Pluripotent Stem Cells" Genes 14, no. 3: 730. https://doi.org/10.3390/genes14030730