
A Biallelic Truncating Variant in the TPR Domain of GEMIN5 Associated with Intellectual Disability and Cerebral Atrophy
 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Molecular Investigation
2.2. Protein Model
3. Results
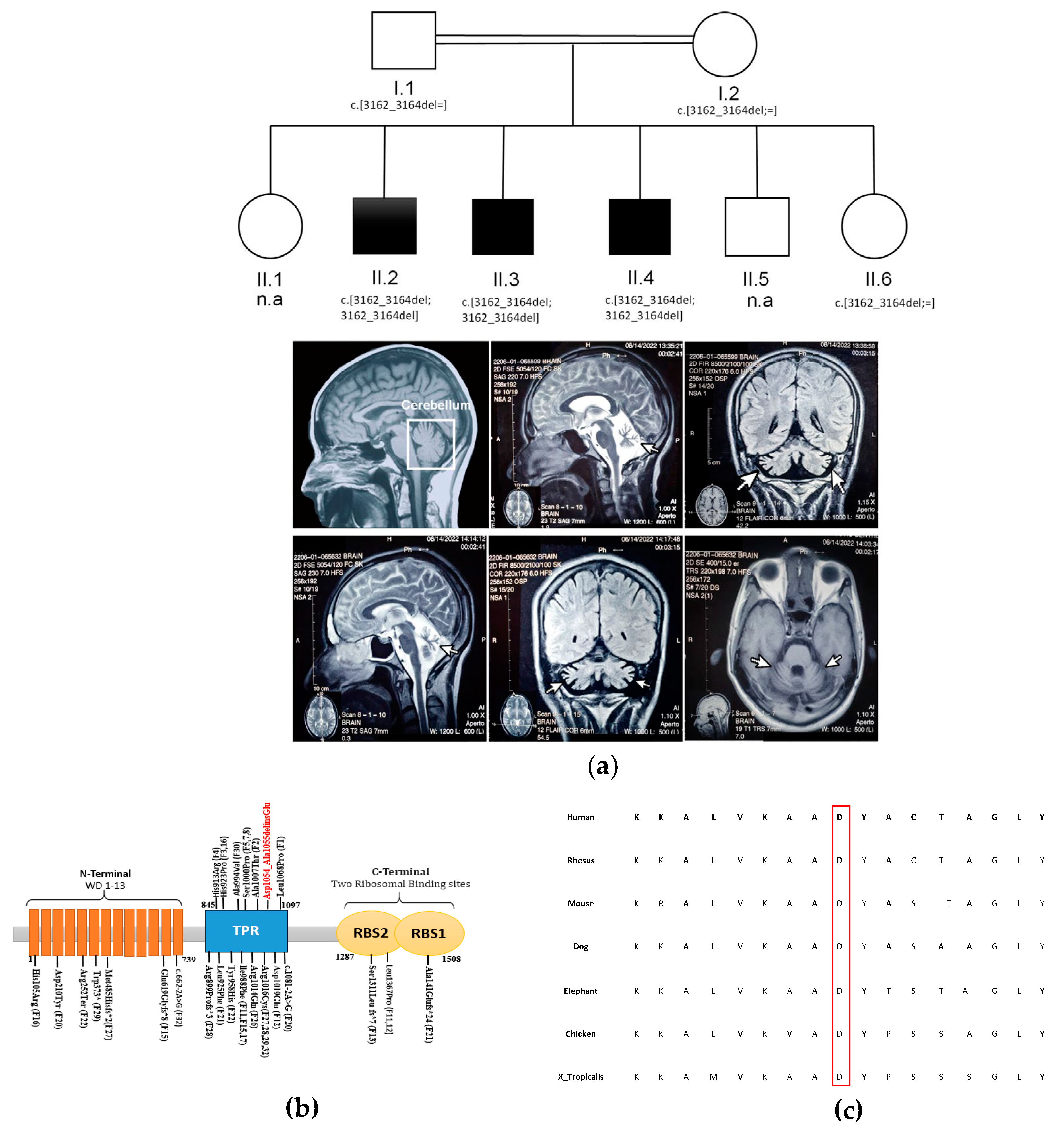
3.1. Clinical Description
3.2. WES Analysis
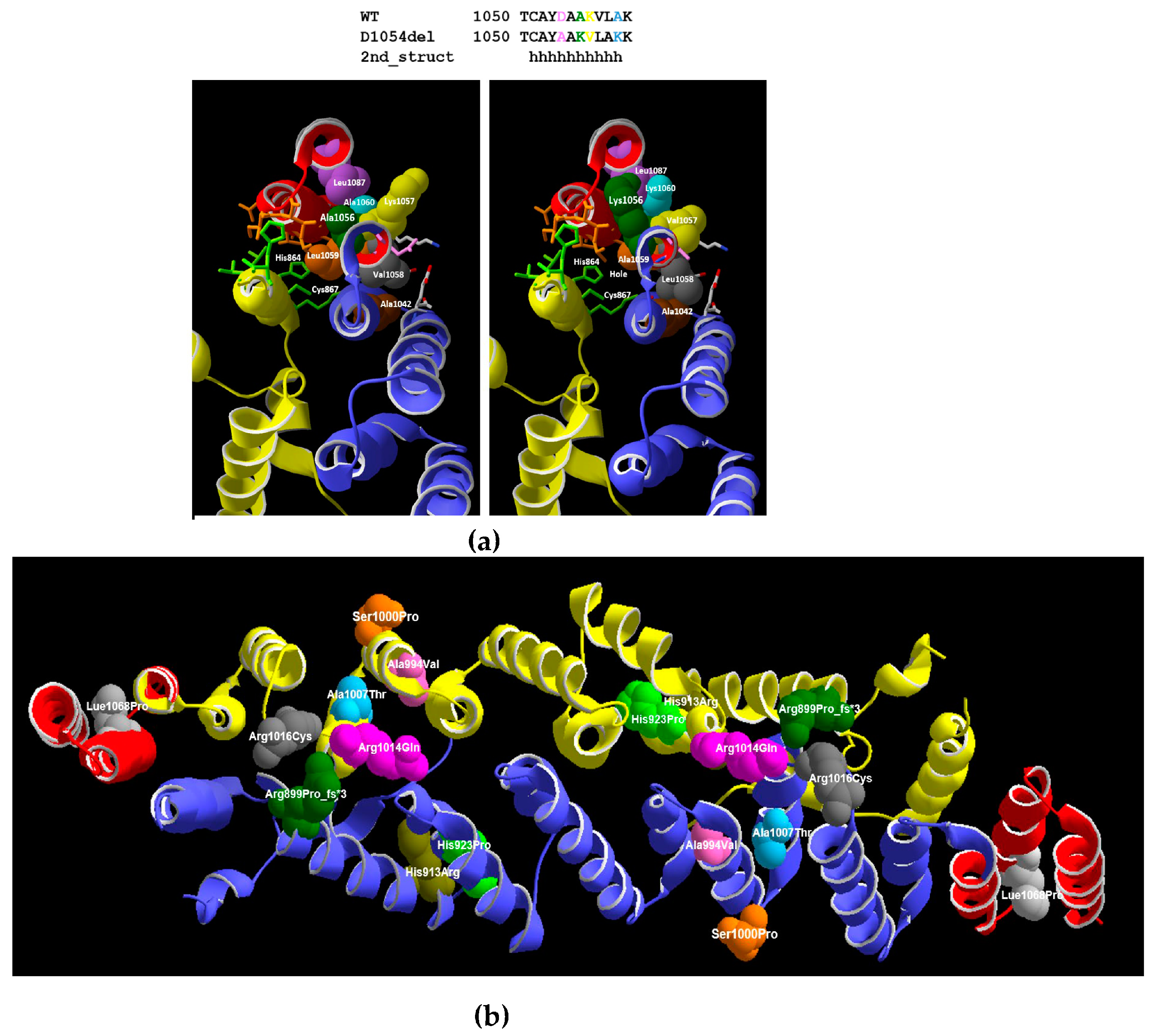
3.3. 3D Model
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bosia, M.; Seghi, F.; Bigai, G.; Martini, F.; Fregna, L.; Fazio, V.; Cavallaro, R. Adult Consequences of Neurodevelopmental Disorders. In Fundamentals of Psychiatry for Health Care Professionals; Springer International Publishing: Cham, Germany, 2022; pp. 199–227. [Google Scholar]
- Bruno, L.P.; Doddato, G.; Valentino, F.; Baldassarri, M.; Tita, R.; Fallerini, C.; Bruttini, M.; Lo Rizzo, C.; Mencarelli, M.A.; Mari, F.; et al. New Candidates for Autism/Intellectual Disability Identified by Whole-Exome Sequencing. Int. J. Mol. Sci. 2021, 22, 13439. [Google Scholar] [CrossRef] [PubMed]
- Harripaul, R.; Vasli, N.; Mikhailov, A.; Rafiq, M.A.; Mittal, K.; Windpassinger, C.; Sheikh, T.I.; Noor, A.; Mahmood, H.; Downey, S.; et al. Mapping Autosomal Recessive Intellectual Disability: Combined Microarray and Exome Sequencing Identifies 26 Novel Candidate Genes in 192 Consanguineous Families. Mol. Psychiatry 2018, 23, 973–984. [Google Scholar] [CrossRef] [PubMed]
- Ropers, H.H. Genetics of Early Onset Cognitive Impairment. Annu. Rev. Genomics Hum. Genet. 2010, 11, 161–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilissen, C.; Hehir-Kwa, J.Y.; Thung, D.T.; van de Vorst, M.; van Bon, B.W.M.; Willemsen, M.H.; Kwint, M.; Janssen, I.M.; Hoischen, A.; Schenck, A.; et al. Genome Sequencing Identifies Major Causes of Severe Intellectual Disability. Nature 2014, 511, 344–347. [Google Scholar] [CrossRef]
- Riazuddin, S.; Hussain, M.; Razzaq, A.; Iqbal, Z.; Shahzad, M.; Polla, D.L.; Song, Y.; van Beusekom, E.; Khan, A.A.; Tomas-Roca, L.; et al. Exome Sequencing of Pakistani Consanguineous Families Identifies 30 Novel Candidate Genes for Recessive Intellectual Disability. Mol. Psychiatry 2017, 22, 1604–1614. [Google Scholar] [CrossRef] [Green Version]
- Antonarakis, S.E. Carrier Screening for Recessive Disorders. Nat. Rev. Genet. 2019, 20, 549–561. [Google Scholar] [CrossRef]
- Ilyas, M.; Mir, A.; Efthymiou, S.; Houlden, H. The Genetics of Intellectual Disability: Advancing Technology and Gene Editing. F1000Research 2020, 9, 22. [Google Scholar] [CrossRef] [Green Version]
- Iqbal, S.; Zakar, R.; Fischer, F.; Zakar, M.Z. Consanguineous Marriages and Their Association with Women’s Reproductive Health and Fertility Behavior in Pakistan: Secondary Data Analysis from Demographic and Health Surveys, 1990–2018. BMC Womens. Health 2022, 22, 118. [Google Scholar] [CrossRef]
- Naz, S.; Ibrahim, N.; Sharif, S.; Bashir, N.; Sajjad, E.; Asghar, I.; Irshad, S.; Firasat, S.; Kaul, H.; Sarwar, S. Prevalence and Association of Different Levels of Intellectual Disability with Prenatal, Perinatal, Neonatal and Postnatal Factors. Proc. Pakistan Acad. Sci. B. Life Environ. Sci. 2022, 58, 75–82. [Google Scholar] [CrossRef]
- Müller-McNicoll, M.; Neugebauer, K.M. How Cells Get the Message: Dynamic Assembly and Function of MRNA–Protein Complexes. Nat. Rev. Genet. 2013, 14, 275–287. [Google Scholar] [CrossRef]
- Jonas, K.; Calin, G.A.; Pichler, M. RNA-Binding Proteins as Important Regulators of Long Non-Coding RNAs in Cancer. Int. J. Mol. Sci. 2020, 21, 2969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francisco-Velilla, R.; Embarc-Buh, A.; del Caño-Ochoa, F.; Abellan, S.; Vilar, M.; Alvarez, S.; Fernandez-Jaen, A.; Kour, S.; Rajan, D.S.; Pandey, U.B.; et al. Functional and Structural Deficiencies of Gemin5 Variants Associated with Neurological Disorders. Life Sci. Alliance 2022, 5, e202201403. [Google Scholar] [CrossRef] [PubMed]
- Gubitz, A. The SMN Complex. Exp. Cell Res. 2004, 296, 51–56. [Google Scholar] [CrossRef]
- Piazzon, N.; Schlotter, F.; Lefebvre, S.; Dodre, M.; Mereau, A.; Soret, J.; Besse, A.; Barkats, M.; Bordonne, R.; Branlant, C.; et al. Implication of the SMN Complex in the Biogenesis and Steady State Level of the Signal Recognition Particle. Nucleic Acids Res. 2013, 41, 1255–1272. [Google Scholar] [CrossRef] [Green Version]
- Francisco-Velilla, R.; Fernandez-Chamorro, J.; Ramajo, J.; Martinez-Salas, E. The RNA-Binding Protein Gemin5 Binds Directly to the Ribosome and Regulates Global Translation. Nucleic Acids Res. 2016, 44, 8335–8351. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Salas, E.; Embarc-Buh, A.; Francisco-Velilla, R. Emerging Roles of Gemin5: From SnRNPs Assembly to Translation Control. Int. J. Mol. Sci. 2020, 21, 3868. [Google Scholar] [CrossRef]
- Embarc-Buh, A.; Francisco-Velilla, R.; Camero, S.; Pérez-Cañadillas, J.M.; Martínez-Salas, E. The RBS1 Domain of Gemin5 Is Intrinsically Unstructured and Interacts with RNA through Conserved Arg and Aromatic Residues. RNA Biol. 2021, 18, 496–506. [Google Scholar] [CrossRef]
- Moreno-Morcillo, M.; Francisco-Velilla, R.; Embarc-Buh, A.; Fernández-Chamorro, J.; Ramón-Maiques, S.; Martinez-Salas, E. Structural Basis for the Dimerization of Gemin5 and Its Role in Protein Recruitment and Translation Control. Nucleic Acids Res. 2020, 48, 788–801. [Google Scholar] [CrossRef] [Green Version]
- Francisco-Velilla, R.; Azman, E.; Martinez-Salas, E. Impact of RNA–Protein Interaction Modes on Translation Control: The Versatile Multidomain Protein Gemin5. BioEssays 2019, 41, 1800241. [Google Scholar] [CrossRef]
- Piñeiro, D.; Fernandez-Chamorro, J.; Francisco-Velilla, R.; Martinez-Salas, E. Gemin5: A Multitasking RNA-Binding Protein Involved in Translation Control. Biomolecules 2015, 5, 528–544. [Google Scholar] [CrossRef] [Green Version]
- Piñeiro, D.; Fernández, N.; Ramajo, J.; Martínez-Salas, E. Gemin5 Promotes IRES Interaction and Translation Control through Its C-terminal Region. Nucleic Acids Res. 2013, 41, 1017–1028. [Google Scholar] [CrossRef] [PubMed]
- Rajan, D.S.; Kour, S.; Fortuna, T.R.; Cousin, M.A.; Barnett, S.S.; Niu, Z.; Babovic-Vuksanovic, D.; Klee, E.W.; Kirmse, B.; Innes, M.; et al. Autosomal Recessive Cerebellar Atrophy and Spastic Ataxia in Patients With Pathogenic Biallelic Variants in GEMIN5. Front. Cell Dev. Biol. 2022, 10, 10. [Google Scholar] [CrossRef] [PubMed]
- Kour, S.; Rajan, D.S.; Fortuna, T.R.; Anderson, E.N.; Ward, C.; Lee, Y.; Lee, S.; Shin, Y.B.; Chae, J.-H.; Choi, M.; et al. Loss of Function Mutations in GEMIN5 Cause a Neurodevelopmental Disorder. Nat. Commun. 2021, 12, 2558. [Google Scholar] [CrossRef]
- Saida, K.; Tamaoki, J.; Sasaki, M.; Haniffa, M.; Koshimizu, E.; Sengoku, T.; Maeda, H.; Kikuchi, M.; Yokoyama, H.; Sakamoto, M.; et al. Pathogenic Variants in the Survival of Motor Neurons Complex Gene GEMIN5 Cause Cerebellar Atrophy. Clin. Genet. 2021, 100, 722–730. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, A.; Naz, S.; Kaul, H.; Sharif, S.; Khushbakht, A.; Naeem, M.A.; Iqtedar, M.; Kaleem, A.; Firasat, S.; Manzoor, F. Mutational Analysis in Sodium-Borate Cotransporter SLC4A11 in Consanguineous Families from Punjab, Pakistan. PLoS ONE 2022, 17, e0273685. [Google Scholar] [CrossRef]
- Grimberg, J.; Nawoschik, S.; Belluscio, L.; McKee, R.; Turck, A.; Eisenberg, A. A Simple and Efficient Non-Organic Procedure for the Isolation of Genomic DNA from Blood. Nucleic Acids Res. 1989, 17, 8390. [Google Scholar] [CrossRef]
- Mattioli, F.; Darvish, H.; Paracha, S.A.; Tafakhori, A.; Firouzabadi, S.G.; Chapi, M.; Baig, H.M.A.; Reymond, A.; Antonarakis, S.E.; Ansar, M. Biallelic Truncation Variants in ATP9A Are Associated with a Novel Autosomal Recessive Neurodevelopmental Disorder. NPJ Genomic Med. 2021, 6, 94. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows–Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce Framework for Analyzing next-Generation DNA Sequencing Data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Delafontaine, J.; Masselot, A.; Liechti, R.; Kuznetsov, D.; Xenarios, I.; Pradervand, S. Varapp: A Reactive Web-Application for Variants Filtering. bioRxiv 2016, 060806. [Google Scholar] [CrossRef] [Green Version]
- Johansson, M.U.; Zoete, V.; Michielin, O.; Guex, N. Defining and Searching for Structural Motifs Using DeepView/Swiss-PdbViewer. BMC Bioinformatics 2012, 13, 173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Shree, A.; Shukla, P.C. Intellectual Disability: Definition, Classification, Causes and Characteristics. Learn. Community-An Int. J. Educ. Soc. Dev. 2016, 7, 9. [Google Scholar] [CrossRef]
- Gubitz, A.K.; Mourelatos, Z.; Abel, L.; Rappsilber, J.; Mann, M.; Dreyfuss, G. Gemin5, a Novel WD Repeat Protein Component of the SMN Complex That Binds Sm Proteins. J. Biol. Chem. 2002, 277, 5631–5636. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Chamorro, J.; Piñeiro, D.; Gordon, J.M.B.; Ramajo, J.; Francisco-Velilla, R.; Macias, M.J.; Martinez-Salas, E. Identification of Novel Non-Canonical RNA-Binding Sites in Gemin5 Involved in Internal Initiation of Translation. Nucleic Acids Res. 2014, 42, 5742–5754. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Phenotypes of Patients with Biallelic GEMIN5 Variants [13,23,24,25] and Our Study | Our Study | Phenotypes of Patients with Variants in the TPR Domains [13,23,24] and Our Study | Phenotypes of Patients with Variants Outside the TPR Domains [13,23,24,25] | |||
|---|---|---|---|---|---|---|
| Total Patients | 45 | II.2 | II.3 | II.4 | 17 | 15 |
| Sex; male/female (M/F) | 69% M (31) 31% F (14) | M | M | M | 59% (10) M, 41% (7) F | 73% (11) M, 27% (4) F |
| Onset | <1 | After 5 years | <1 | |||
| Age at last evaluation | 46 Y | 35 Y | 28 Y | |||
| Origin | Pakistani | Pakistani | Pakistani | |||
| Language | Punjabi | Punjabi | Punjabi | |||
| Consanguineous parents (Number of families for yes) | 6/28 (21%) | + | + | + | 5/10 (50%) | 0/7 (0%) |
| Genetic testing | ||||||
| Kind of Variant | Homozygous inframe deletion | Homozygous inframe deletion | Homozygous inframe deletion | |||
| Chromosome number: Transcript | Ch5: (NM_015465.5; NP_056280.2) | Ch5: (NM_015465.5; NP_056280.2) | Ch5: (NM_015465.5; NP_056280.2) | |||
| Variant (g. DNA; protein) | c.3162_3164del; p. (Asp1054_Ala1055delinsGlu) | c.3162_3164del; p. (Asp1054_Ala1055delinsGlu) | c.3162_3164de; p. (Asp1054_Ala1055delinsGlu) | |||
| Growth at last investigation | ||||||
| Height (cm) | 159.6 | 161.5 | 167.6 | |||
| Weight (kg) | 40 | 54 | 56 | |||
| Head circumference (cm) | 51 | 53.3 | 53.3 | |||
| Development | ||||||
| Delayed | 96% (43/45) | + | + | + | 94% (16/17) | 93% (14/15) |
| Cognitive delay | 88% (37/42) | + | + | + | 100% (15/15) | 73% (11/15) |
| Intellectual disability (Mild to severe) | 98% (41/42) | Severe | Mild | Mild | 100% (15/15) | 93% (14/15) |
| Motor delay | 95% (41/43) | + | - | + | 88% (15/17) | 94% (12/13) |
| Speech delay | 95% (39/41) | + | - | + | 93% (13/14) | 92% (14/14) |
| Regression (−) | 100% (42/42) | - | - | - | 100% (16/16) | 100% (13/13) |
| Neurological finding | ||||||
| Ataxia | 74% (31/42) | + | - | + | 60% (9/15) | 79% (11/14) |
| Walking difficulties | 95% (40/42) | + | - | + | 87% (13/15) | 100% (14/14) |
| Neurological Evaluation | ||||||
| MRI Scans | 100% (44/44) | Diffused Cerebral atrophy | Not done | Mild diffuse cerebral atrophy | 100% (16/16) | 100% (15/15) |
| Behavioral anomalies | ||||||
| Aggressive behavior | + | - | + | |||
| Seizures | - | + | ||||
| Clinical Course | ||||||
| Progressive/static (P/S) | 78% S (29/37) 22% P (8/37) | P | S | S | 86% (12) S, 14% (2) P | 83% (10) S, 17% (2) P |
| Others | Sinus mucosal disease, no maleness | Sinus mucosal disease | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ibrahim, N.; Naz, S.; Mattioli, F.; Guex, N.; Sharif, S.; Iqbal, A.; Ansar, M.; Reymond, A. A Biallelic Truncating Variant in the TPR Domain of GEMIN5 Associated with Intellectual Disability and Cerebral Atrophy. Genes 2023, 14, 707. https://doi.org/10.3390/genes14030707
Ibrahim N, Naz S, Mattioli F, Guex N, Sharif S, Iqbal A, Ansar M, Reymond A. A Biallelic Truncating Variant in the TPR Domain of GEMIN5 Associated with Intellectual Disability and Cerebral Atrophy. Genes. 2023; 14(3):707. https://doi.org/10.3390/genes14030707
Chicago/Turabian StyleIbrahim, Nazia, Shagufta Naz, Francesca Mattioli, Nicolas Guex, Saima Sharif, Afia Iqbal, Muhammad Ansar, and Alexandre Reymond. 2023. "A Biallelic Truncating Variant in the TPR Domain of GEMIN5 Associated with Intellectual Disability and Cerebral Atrophy" Genes 14, no. 3: 707. https://doi.org/10.3390/genes14030707