
High Performance of a Dominant/X-Linked Gene Panel in Patients with Neurodevelopmental Disorders
 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients Description and Classification
2.2. Gene Panel Content
2.3. Sequencing and Data Processing
2.4. Variant Filtering
2.5. Re-Analysis Variant Filtering
2.6. Copy Number Variants Detection and Filtering
2.7. Exome Sequencing
2.8. Variant Validation and Classification
3. Results
3.1. Diagnostic Yield, Variant Types, and Genes
3.2. Data Re-Analysis
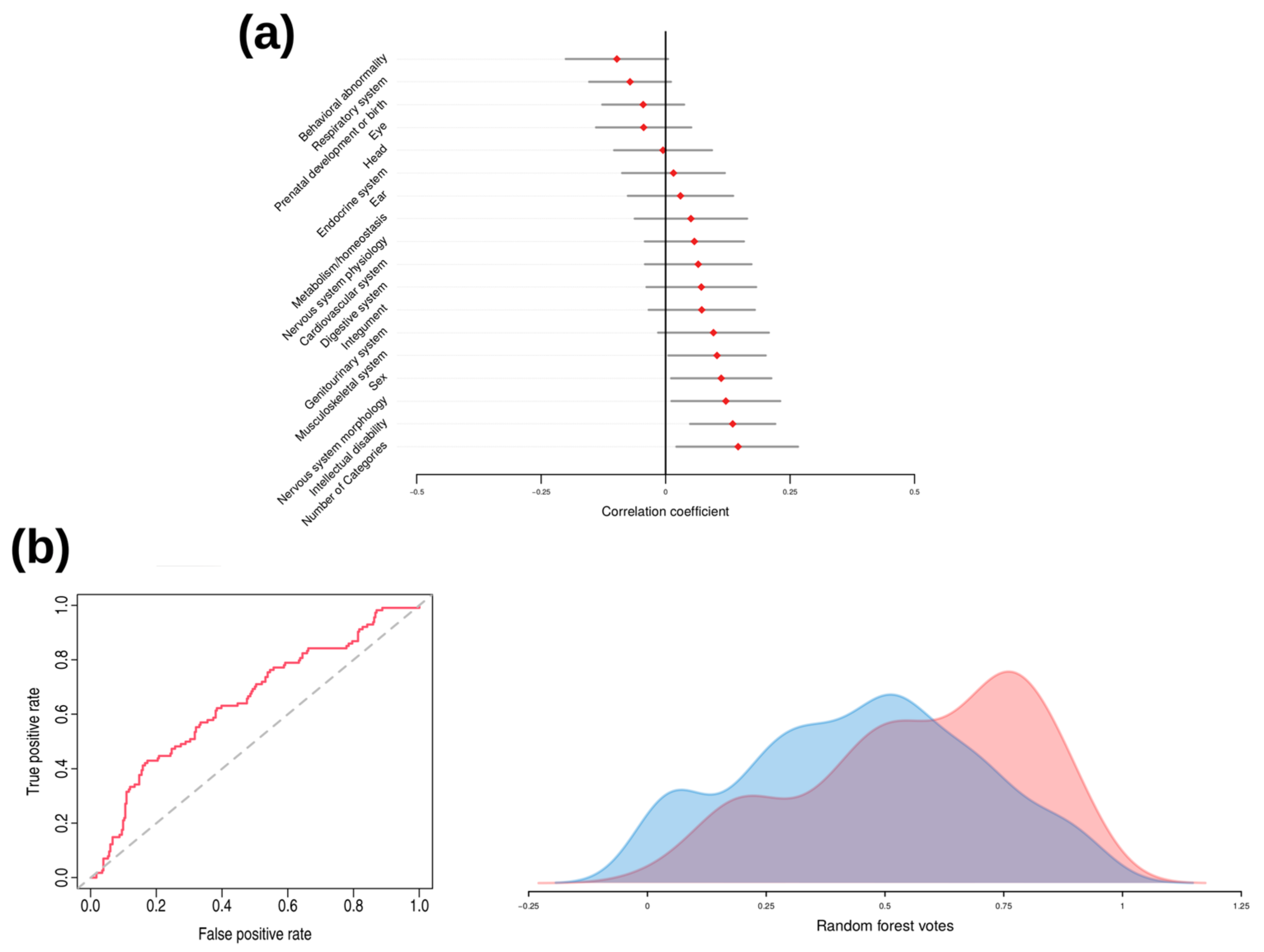
3.3. Sex and Patient Subset Analysis
3.4. Clinical Profiling
3.5. WES Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Boyle, C.A.; Boulet, S.; Schieve, L.A.; Cohen, R.A.; Blumberg, S.J.; Yeargin-Allsopp, M.; Visser, S.; Kogan, M.D. Trends in the prevalence of developmental disabilities in US children, 1997–2008. Pediatrics 2011, 127, 1034–1042. [Google Scholar] [CrossRef]
- Perou, R.; Bitsko, R.H.; Blumberg, S.J.; Pastor, P.; Ghandour, R.M.; Gfroerer, J.C.; Hedden, S.L.; Crosby, A.E.; Visser, S.N.; Schieve, L.A.; et al. Mental health surveillance among children—United States, 2005–2011. MMWR Suppl. 2013, 62, 1–35. [Google Scholar] [PubMed]
- DSM Library. Diagnostic and Statistical Manual of Mental Disorders. Available online: https://dsm.psychiatryonline.org/doi/book/10.1176/appi.books.9780890425596 (accessed on 4 January 2023).
- Kaufman, L.; Ayub, M.; Vincent, J.B. The genetic basis of non-syndromic intellectual disability: A review. J. Neurodev. Disord. 2010, 2, 182–209. [Google Scholar] [CrossRef] [PubMed]
- May, M.E.; Kennedy, C.H. Health and problem behavior among people with intellectual disabilities. Behav. Anal. Pract. 2010, 3, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Chiurazzi, P.; Pirozzi, F. Advances in understanding—Genetic basis of intellectual disability. F1000Research 2016, 5, 599. [Google Scholar] [CrossRef]
- Manickam, K.; McClain, M.R.; Demmer, L.A.; Biswas, S.; Kearney, H.M.; Malinowski, J.; Massingham, L.J.; Miller, D.; Yu, T.W.; Hisama, F.M. Exome and genome sequencing for pediatric patients with congenital anomalies or intellectual disability: An evidence-based clinical guideline of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2021, 23, 2029–2037. [Google Scholar] [CrossRef] [PubMed]
- Wright, C.F.; McRae, J.F.; Clayton, S.; Gallone, G.; Aitken, S.; FitzGerald, T.W.; Jones, P.; Prigmore, E.; Rajan, D.; Lord, J.; et al. Making new genetic diagnoses with old data: Iterative reanalysis and reporting from genome-wide data in 1133 families with developmental disorders. Genet. Med. 2018, 20, 1216–1223. [Google Scholar] [CrossRef]
- Stefanski, A.; Calle-López, Y.; Leu, C.; Pérez-Palma, E.; Pestana-Knight, E.; Lal, D. Clinical sequencing yield in epilepsy, autism spectrum disorder, and intellectual disability: A systematic review and meta-analysis. Epilepsia 2021, 62, 143–151. [Google Scholar] [CrossRef]
- Abrahams, B.S.; E Arking, D.; Campbell, D.B.; Mefford, H.C.; Morrow, E.M.; A Weiss, L.; Menashe, I.; Wadkins, T.; Banerjee-Basu, S.; Packer, A. SFARI Gene 2.0: A community-driven knowledgebase for the autism spectrum disorders (ASDs). Mol. Autism. 2013, 4, 36. [Google Scholar] [CrossRef]
- Kochinke, K.; Zweier, C.; Nijhof, B.; Fenckova, M.; Cizek, P.; Honti, F.; Keerthikumar, S.; Oortveld, M.A.; Kleefstra, T.; Kramer, J.M.; et al. Systematic Phenomics Analysis Deconvolutes Genes Mutated in Intellectual Disability into Biologically Coherent Modules. Am. J. Hum. Genet. 2016, 98, 149–164. [Google Scholar] [CrossRef]
- Jensen, M.; Girirajan, S. Mapping a shared genetic basis for neurodevelopmental disorders. Genome Med. 2017, 9, 109. [Google Scholar] [CrossRef]
- Srivastava, A.K.; Schwartz, C.E. Intellectual disability and autism spectrum disorders: Causal genes and molecular mechanisms. Neurosci. Biobehav. Rev. 2014, 46 Pt 2, 161–174. [Google Scholar] [CrossRef]
- Parenti, I.; Rabaneda, L.G.; Schoen, H.; Novarino, G. Neurodevelopmental Disorders: From Genetics to Functional Pathways. Trends Neurosci. 2020, 43, 608–621. [Google Scholar] [CrossRef] [PubMed]
- Dulac, C. Brain function and chromatin plasticity. Nature 2010, 465, 728–735. [Google Scholar] [CrossRef]
- Han, J.Y.; Lee, I.G. Genetic tests by next-generation sequencing in children with developmental delay and/or intellectual disability. Clin. Exp. Pediatr. 2020, 63, 195–202. [Google Scholar] [CrossRef]
- Bruel, A.-L.; Vitobello, A.; Mau-Them, F.T.; Nambot, S.; Sorlin, A.; Denommé-Pichon, A.; Delanne, J.; Moutton, S.; Callier, P.; Duffourd, Y.; et al. Next-generation sequencing approaches and challenges in the diagnosis of developmental anomalies and intellectual disability. Clin. Genet. 2020, 98, 433–444. [Google Scholar] [CrossRef]
- Köhler, S.; Gargano, M.; Matentzoglu, N.; Carmody, L.C.; Lewis-Smith, D.; Vasilevsky, N.A.; Danis, D.; Balagura, G.; Baynam, G.; Brower, A.M.; et al. The Human Phenotype Ontology in 2021. Nucleic Acids Res. 2021, 49, D1207. [Google Scholar] [CrossRef] [PubMed]
- Wright, C.F.; Fitzgerald, T.W.; Jones, W.D.; Clayton, S.; McRae, J.F.; van Kogelenberg, M.; King, D.A.; Ambridge, K.; Barrett, D.M.; Bayzetinova, T.; et al. Genetic diagnosis of developmental disorders in the DDD study: A scalable analysis of genome-wide research data. Lancet 2015, 385, 1305–1314. [Google Scholar] [CrossRef]
- de Sena Brandine, G.; Smith, A.D. Falco: High-speed FastQC emulation for quality control of sequencing data. F1000Research 2019, 8, 1874. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef] [PubMed]
- Poplin, R.; Ruano-Rubio, V.; DePristo, M.A.; Fennell, T.J.; Carneiro, M.O.; Van der Auwera, G.A.; Kling, D.E.; Gauthier, L.D.; Levy-Moonshine, A.; Roazen, D.; et al. Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv 2018, 201178. [Google Scholar] [CrossRef]
- Liu, X.; White, S.; Peng, B.; Johnson, A.D.; A Brody, J.; Li, A.H.; Huang, Z.; Carroll, A.; Wei, P.; Gibbs, R.; et al. WGSA: An annotation pipeline for human genome sequencing studies. J. Med. Genet. 2016, 53, 111–112. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alfoldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Danis, D.; Jacobsen, J.O.; Carmody, L.C.; Gargano, M.A.; McMurry, J.A.; Hegde, A.; Haendel, M.A.; Valentini, G.; Smedley, D.; Robinson, P.N. Interpretable prioritization of splice variants in diagnostic next-generation sequencing. Am. J. Hum. Genet. 2021, 108, 1564–1577. [Google Scholar] [CrossRef]
- Jaganathan, K.; Panagiotopoulou, S.K.; McRae, J.F.; Darbandi, S.F.; Knowles, D.; Li, Y.I.; Kosmicki, J.A.; Arbelaez, J.; Cui, W.; Schwartz, G.B.; et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell 2019, 176, 535–548.e24. [Google Scholar] [CrossRef]
- Jian, X.; Boerwinkle, E.; Liu, X. In silico prediction of splice-altering single nucleotide variants in the human genome. Nucleic Acids Res. 2014, 42, 13534–13544. [Google Scholar] [CrossRef]
- Choi, Y.; Chan, A.P. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015, 31, 2745–2747. [Google Scholar] [CrossRef]
- Fromer, M.; Purcell, S.M. Using XHMM Software to Detect Copy Number Variation in Whole-Exome Sequencing Data. Curr Protoc. Hum. Genet. 2014, 81, 7.23.1–7.23.21. [Google Scholar] [CrossRef]
- Plagnol, V.; Curtis, J.; Epstein, M.; Mok, K.Y.; Stebbings, E.; Grigoriadou, S.; Wood, N.W.; Hambleton, S.; Burns, S.O.; Thrasher, A.J.; et al. A robust model for read count data in exome sequencing experiments and implications for copy number variant calling. Bioinformatics 2012, 28, 2747–2754. [Google Scholar] [CrossRef]
- MacDonald, J.R.; Ziman, R.; Yuen, R.K.C.; Feuk, L.; Scherer, S.W. The Database of Genomic Variants: A curated collection of structural variation in the human genome. Nucleic Acids Res. 2014, 42, D986–D992. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Tohyama, J.; Kato, M.; Akasaka, N.; Magara, S.; Kawashima, H.; Ohashi, T.; Shiraishi, H.; Nakashima, M.; Saitsu, H.; et al. High prevalence of genetic alterations in early-onset epileptic encephalopathies associated with infantile movement disorders. Brain Dev. 2016, 38, 285–292. [Google Scholar] [CrossRef]
- Hoyer, J.; Ekici, A.B.; Endele, S.; Popp, B.; Zweier, C.; Wiesener, A.; Wohlleber, E.; Dufke, A.; Rossier, E.; Petsch, C.; et al. Haploinsufficiency of ARID1B, a member of the SWI/SNF-a chromatin-remodeling complex, is a frequent cause of intellectual disability. Am. J. Hum. Genet. 2012, 90, 565–572. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Cayuelas, E.; Blanco-Kelly, F.; Lopez-Grondona, F.; Swafiri, S.T.; Lopez-Rodriguez, R.; Pozo, R.L.-D.; Mahillo-Fernandez, I.; Moreno, B.; Rodrigo-Moreno, M.; Casas-Alba, D.; et al. Clinical description, molecular delineation and genotype-phenotype correlation in 340 patients with KBG syndrome: Addition of 67 new patients. J. Med. Genet 2022. online first. [Google Scholar] [CrossRef] [PubMed]
- Sitzmann, A.F.; Hagelstrom, R.T.; Tassone, F.; Hagerman, R.J.; Butler, M.G. Rare FMR1 gene mutations causing fragile X syndrome: A review. Am. J. Med. Genet. A 2018, 176, 11–18. [Google Scholar] [CrossRef]
- Shaffer, L.G.; Theisen, A.; Bejjani, B.A.; Ballif, B.C.; Aylsworth, A.S.; Lim, C.; McDonald, M.; Ellison, J.W.; Kostiner, D.; Saitta, S.; et al. The discovery of microdeletion syndromes in the post-genomic era: Review of the methodology and characterization of a new 1q41q42 microdeletion syndrome. Genet. Med. 2007, 9, 607–616. [Google Scholar] [CrossRef]
- Schneider, A.L.; Myers, C.T.; Muir, A.M.; Calvert, S.; Basinger, A.; Perry, M.S.; Rodan, L.; Helbig, K.L.; Chambers, C.; Gorman, K.M.; et al. FBXO28 causes developmental and epileptic encephalopathy with profound intellectual disability. Epilepsia 2021, 62, e13–e21. [Google Scholar] [CrossRef]
- Ghrálaigh, F.N.; McCarthy, E.; Murphy, D.N.; Gallagher, L.; Lopez, L.M. Brief Report: Evaluating the Diagnostic Yield of Commercial Gene Panels in Autism. J. Autism. Dev. Disord. 2023, 53, 484–488. [Google Scholar] [CrossRef]
- Turner, T.N.; Wilfert, A.B.; Bakken, T.E.; Bernier, R.A.; Pepper, M.R.; Zhang, Z.; Torene, R.I.; Retterer, K.; Eichler, E.E. Sex-Based Analysis of De Novo Variants in Neurodevelopmental Disorders. Am. J. Hum. Genet. 2019, 105, 1274–1285. [Google Scholar] [CrossRef]
- Polyak, A.; Rosenfeld, J.A.; Girirajan, S. An assessment of sex bias in neurodevelopmental disorders. Genome Med. 2015, 7, 94. [Google Scholar] [CrossRef] [PubMed]
- Jacquemont, S.; Coe, B.P.; Hersch, M.; Duyzend, M.H.; Krumm, N.; Bergmann, S.; Beckmann, J.S.; Rosenfeld, J.A.; Eichler, E.E. A higher mutational burden in females supports a “female protective model” in neurodevelopmental disorders. Am. J. Hum. Genet. 2014, 94, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Havdahl, A.; Niarchou, M.; Starnawska, A.; Uddin, M.; van der Merwe, C.; Warrier, V. Genetic contributions to autism spectrum disorder. Psychol. Med. 2021, 51, 2260–2273. [Google Scholar] [CrossRef] [PubMed]
- Viñas-Jornet, M.; Esteba-Castillo, S.; Baena, N.; Ribas-Vidal, N.; Ruiz, A.; Torrents-Rodas, D.; Gabau, E.; Vilella, E.; Martorell, L.; Armengol, L.; et al. High Incidence of Copy Number Variants in Adults with Intellectual Disability and Co-morbid Psychiatric Disorders. Behav. Genet. 2018, 48, 323–336. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.S.; Kim, S.Y.; Kim, H.; Hwang, H.; Chae, J.H.; Kim, K.J.; Kim, J.-I.; Lim, B.C. Diagnostic Yield of Epilepsy Panel Testing in Patients With Seizure Onset Within the First Year of Life. Front. Neurol. 2019, 10, 988. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Marmiesse, A.; Roca, I.; Díaz-Flores, F.; Cantarín, V.; Pérez-Poyato, M.S.; Fontalba, A.; Laranjeira, F.; Quintans, S.; Moldovan, O.; Felgueroso, B.; et al. Rare Variants in 48 Genes Account for 42% of Cases of Epilepsy with or without Neurodevelopmental Delay in 246 Pediatric Patients. Front. Neurosci. 2019, 13, 1135. [Google Scholar] [CrossRef]
- Na, J.-H.; Shin, S.; Yang, D.; Kim, B.; Kim, H.D.; Kim, S.; Lee, J.-S.; Choi, J.-R.; Lee, S.-T.; Kang, H.-C. Targeted gene panel sequencing in early infantile onset developmental and epileptic encephalopathy. Brain Dev. 2020, 42, 438–448. [Google Scholar] [CrossRef]
- Zhang, Q.; Li, J.; Zhao, Y.; Bao, X.; Wei, L.; Wang, J. Gene mutation analysis of 175 Chinese patients with early-onset epileptic encephalopathy. Clin. Genet. 2017, 91, 717–724. [Google Scholar] [CrossRef]
- Møller, R.S.; Larsen, L.H.; Johannesen, K.M.; Talvik, I.; Talvik, T.; Vaher, U.; Miranda, M.J.; Farooq, M.; Nielsen, J.E.; Svendsen, L.L.; et al. Gene Panel Testing in Epileptic Encephalopathies and Familial Epilepsies. Mol. Syndromol. 2016, 7, 210–219. [Google Scholar] [CrossRef]


{kind=link}
| ALL | ID/GDD | ASD | ||
|---|---|---|---|---|
| Patients (M/F) | 398 (247/151) | 322 (180/142) | 76 (67/9) | |
| Diagnostic yield | All | 28.6% | 33.2% | 9.2% |
| M | 24.7% | 30.0% | 10.4% | |
| F | 35.1% | 37.3% | 0.0% | |
| Inheritance Pattern | de novo | 73.9% (85) | 64.8% (79) | 85.7% (6) |
| Inherited | 10.4% (12) | 10.2% (11) | 0.9% (1) | |
| X-linked (M/F) | 16.5% (7/12) | 17.6% (7/12) | 0.0% (0) | |
| Uncertain/NA | 15.7% (10/8) | 16.7% (10/8) | 0.0% (0/0) | |
| Variant type | LoF | 53.9% (62) | 51.8% (56) | 85.7% (6) |
| Missense/in frame | 40.0% (41/5) | 41.7% (41/4) | 14.3% (0/1) | |
| Cryptic splicing | 1.8% (2) | 1.9% (2) | 0.0% (0) | |
| CNVs (DEL/DUP) | 4.3% (3/2) | 4.6% (3/2) | 0.0% (0/0) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spataro, N.; Trujillo-Quintero, J.P.; Manso, C.; Gabau, E.; Capdevila, N.; Martinez-Glez, V.; Berenguer-Llergo, A.; Reyes, S.; Brunet, A.; Baena, N.; et al. High Performance of a Dominant/X-Linked Gene Panel in Patients with Neurodevelopmental Disorders. Genes 2023, 14, 708. https://doi.org/10.3390/genes14030708
Spataro N, Trujillo-Quintero JP, Manso C, Gabau E, Capdevila N, Martinez-Glez V, Berenguer-Llergo A, Reyes S, Brunet A, Baena N, et al. High Performance of a Dominant/X-Linked Gene Panel in Patients with Neurodevelopmental Disorders. Genes. 2023; 14(3):708. https://doi.org/10.3390/genes14030708
Chicago/Turabian StyleSpataro, Nino, Juan Pablo Trujillo-Quintero, Carmen Manso, Elisabeth Gabau, Nuria Capdevila, Victor Martinez-Glez, Antoni Berenguer-Llergo, Sara Reyes, Anna Brunet, Neus Baena, and et al. 2023. "High Performance of a Dominant/X-Linked Gene Panel in Patients with Neurodevelopmental Disorders" Genes 14, no. 3: 708. https://doi.org/10.3390/genes14030708