

Evaluation of a Less Invasive Cochlear Implant Surgery in OPA1 Mutations Provoking Deafblindness
 , , ,
, , ,
Abstract
:1. Introduction
1.1. Autosomal Dominant Optic Atrophy
1.2. Cochlear Implantation Surgery for Deafblindness c.1499G>A p.(Arg500His)
2. Materials and Methods
2.1. The Patient
2.2. Audiological Evaluation
2.3. Molecular Analysis
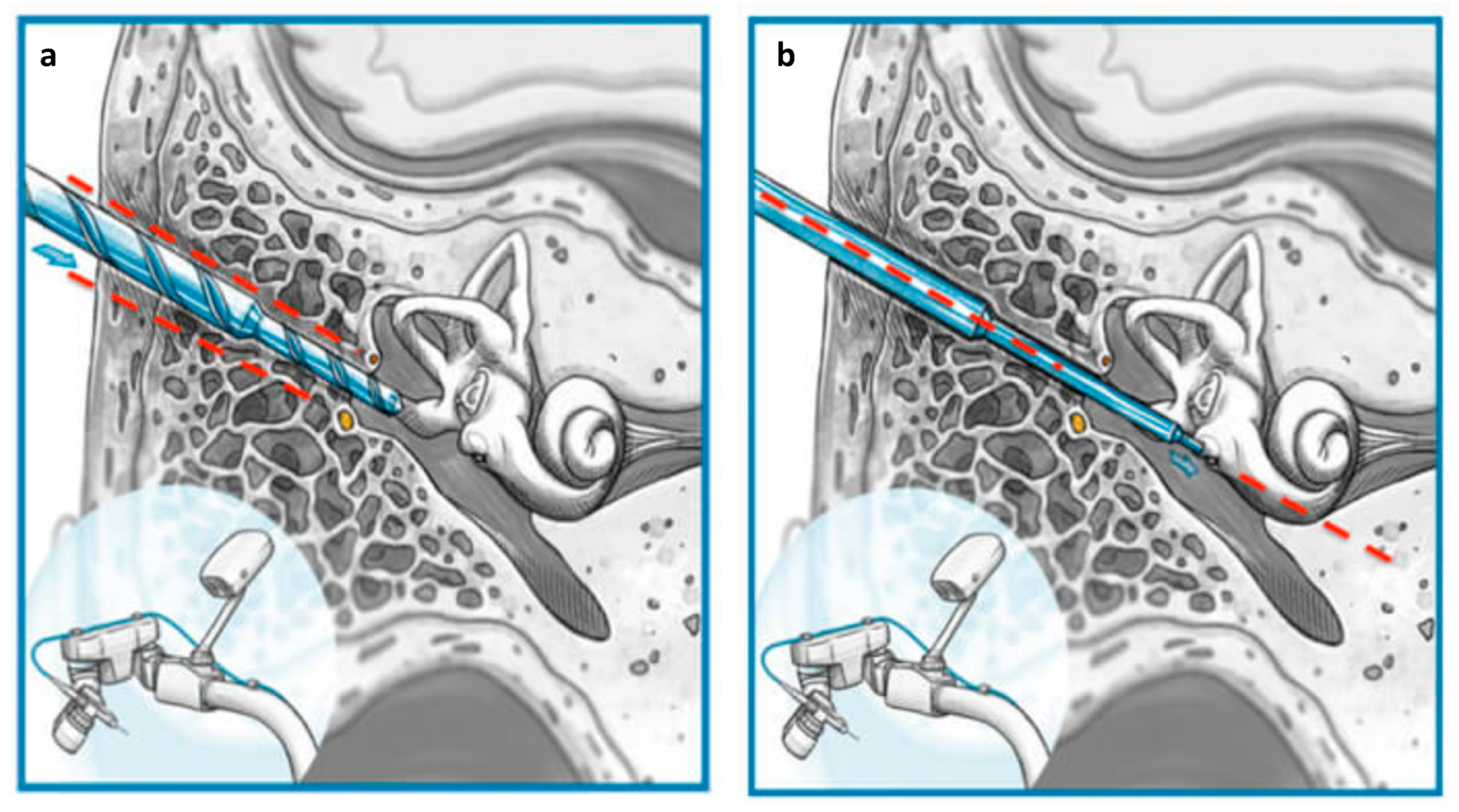
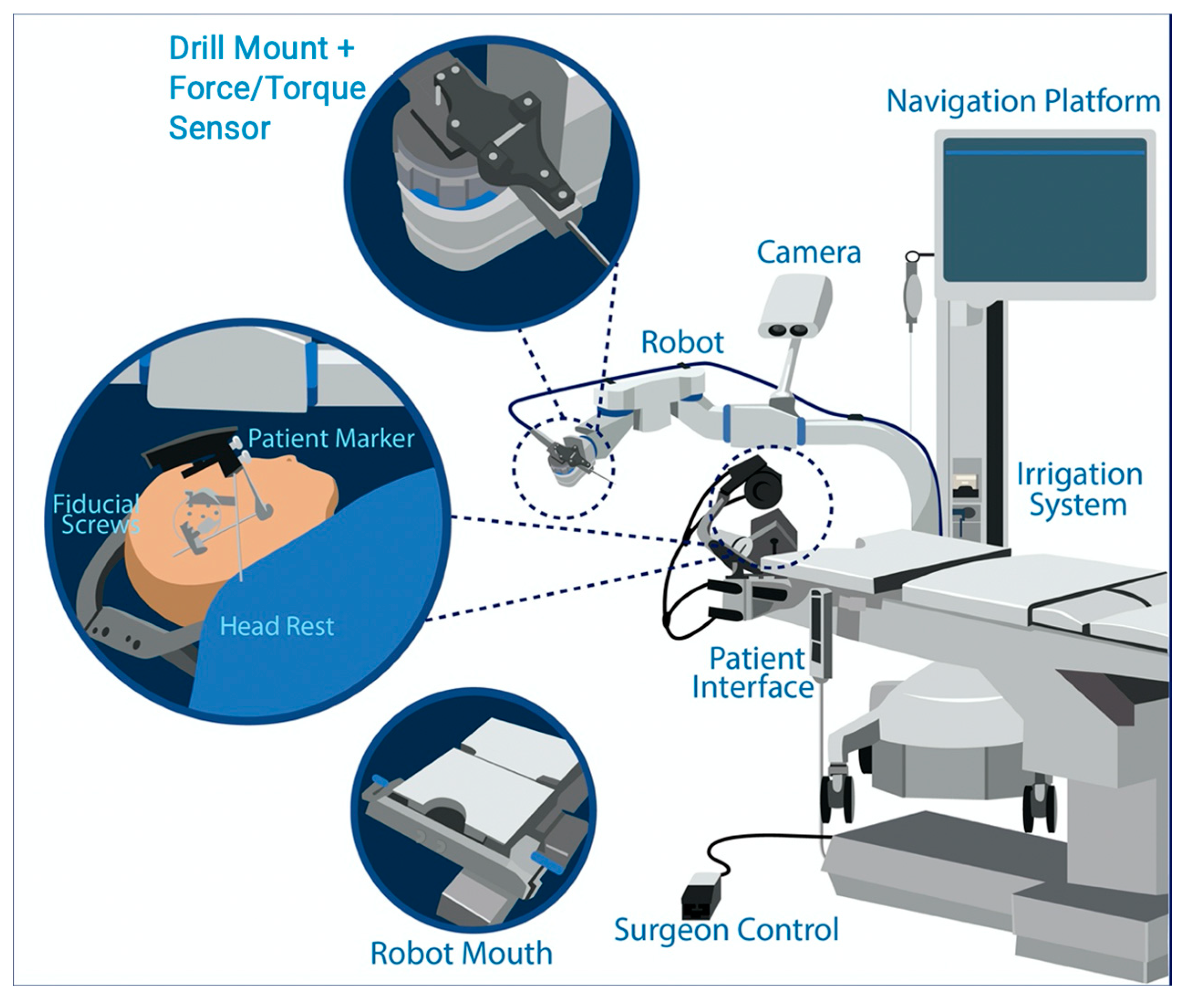
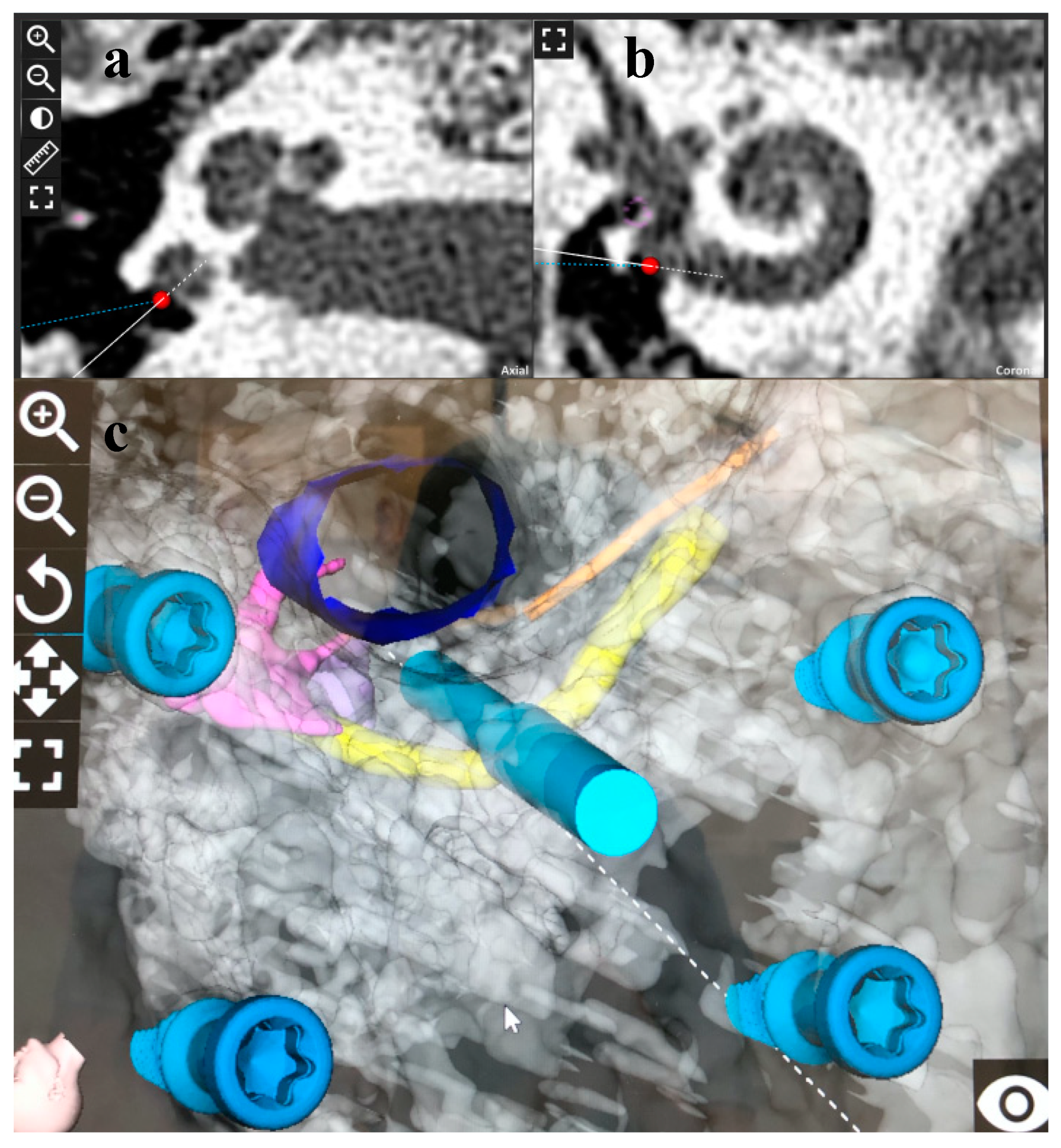
2.4. RACIS: Robotically Assisted Cochlear Implant Surgery
3. Results
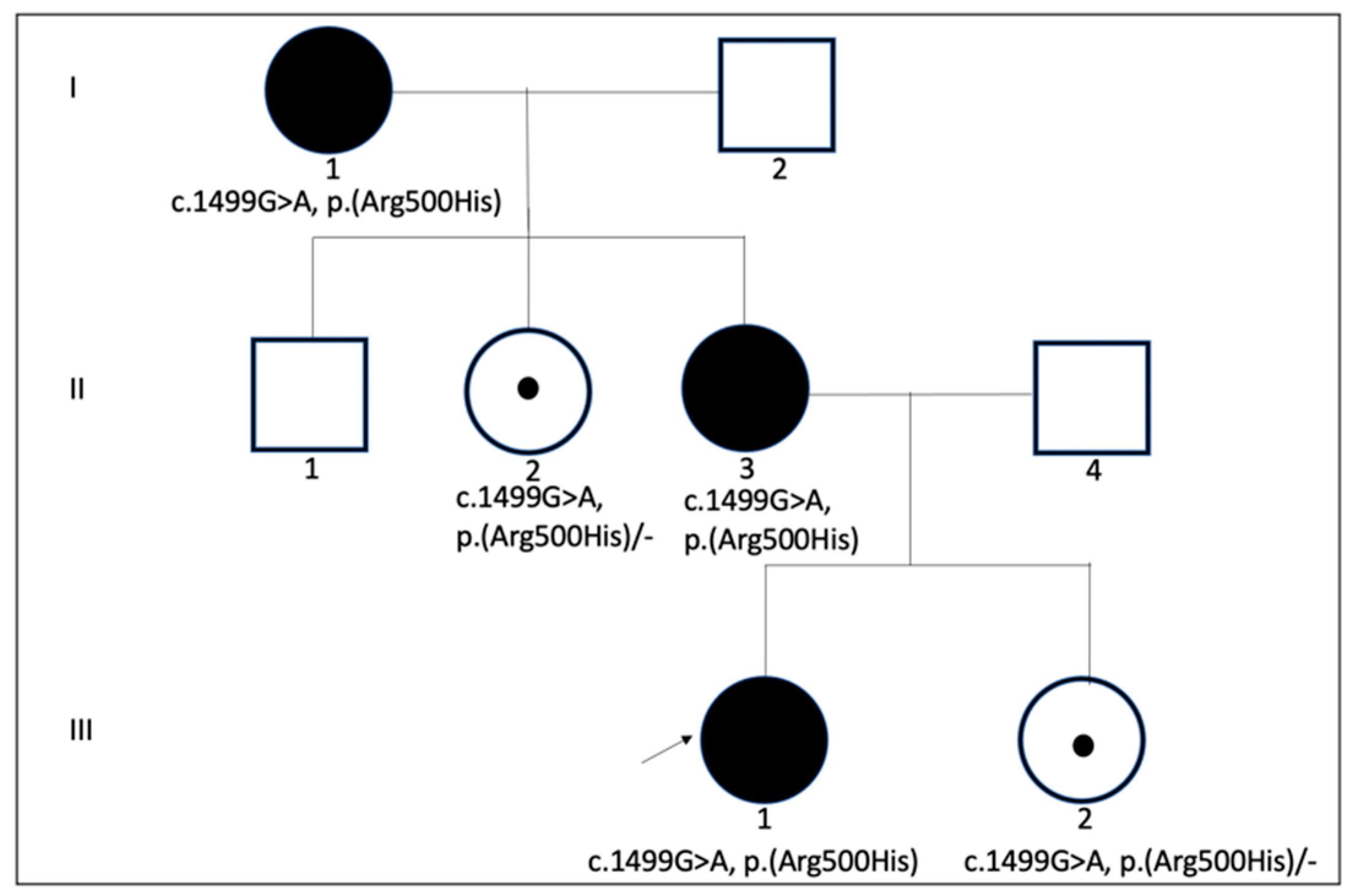
3.1. Molecular Genetic Analysis in OPA1 Mutation and Correlation to Phenotype
3.2. Surgical Results
3.3. Audiological Findings
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- WHO. Available online: https://www.who.int/medicines/areas/priority_medicines/Ch6_21Hearing.pdf (accessed on 15 September 2021).
- D’Haese, P.S.C.; Van Rompaey, V.; De Bodt, M.; Van de Heyning, P. Severe Hearing Loss in the Aging Population Poses a Global Public Health Challenge. How Can We Better Realize the Benefits of Cochlear Implantation to Mitigate This Crisis? Front. Public Health 2019, 7, 227. [Google Scholar] [CrossRef] [Green Version]
- WHO. Available online: https://www.who.int/en/news-room/fact-sheets/detail/dementia (accessed on 15 September 2021).
- Hochman, J.B.; Stockley, T.L.; Shipp, D.; Lin, V.Y.W.; Chen, J.M.; Nedzelski, J.M. Prevalence of Connexin 26 (GJB2) and Pendred (SLC26A4) Mutations in a Population of Adult Cochlear Implant Candidates. Otol. Neurotol. 2010, 31, 919–922. [Google Scholar] [CrossRef]
- Tekin, A.M.; de Ceulaer, G.; Govaerts, P.; Bayazit, Y.; Wuyts, W.; Van de Heyning, P.; Topsakal, V. A New Pathogenic Variant in the TRIOBP Associated with Profound Deafness Is Remediable with Cochlear Implantation. Audiol. Neurotol. 2021, 26, 76–84. [Google Scholar] [CrossRef]
- Morton, C.C.; Nance, W.E. Newborn Hearing Screening—A Silent Revolution. N. Engl. J. Med. 2006, 354, 2151–2164. [Google Scholar] [CrossRef] [PubMed]
- Ehn, M.; Anderzén-Carlsson, A.; Möller, C.; Wahlqvist, M. Life Strategies of People with Deafblindness due to Usher Syndrome Type 2a—A Qualitative Study. Int. J. Qual. Stud. Health Well-Being 2019, 14, 1656790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MEDEL. Available online: https://cdn.shop.medel.com/wp-content-us/uploads/20180613103835/BSN_US-Product-Warranty-and-Service-Contract-Options-Flyer-ENG-and-ESP-Rev.-18.0.pdf (accessed on 16 September 2021).
- Kimura, K.S.; O’Connell, B.P.; Nassiri, A.M.; Dedmon, M.M.; Haynes, D.S.; Bennett, M.L. Outcomes of Revision Cochlear Implantation. Otol. Neurotol. 2020, 41, e705–e711. [Google Scholar] [CrossRef] [PubMed]
- Alexander, C.; Votruba, M.; Pesch, U.E.; Thiselton, D.L.; Mayer, S.; Moore, A.; Rodriguez, M.; Kellner, U.; Leo-Kottler, B.; Auburger, G.; et al. OPA1, Encoding a Dynamin-Related GTPase, Is Mutated in Autosomal Dominant Optic Atrophy Linked to Chromosome 3q28. Nat. Genet. 2000, 26, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Ham, M.; Han, J.; Osann, K.; Smith, M.; Kimonis, V. Meta-Analysis of Genotype-Phenotype Analysis of OPA1 Mutations in Autosomal Dominant Optic Atrophy. Mitochondrion 2019, 46, 262–269. [Google Scholar] [CrossRef]
- Delettre, C.; Lenaers, G.; Pelloquin, L.; Belenguer, P.; Hamel, C.P. OPA1 (Kjer Type) Dominant Optic Atrophy: A Novel Mitochondrial Disease. Mol. Genet. Metab. 2002, 75, 97–107. [Google Scholar] [CrossRef]
- Amati-Bonneau, P.; Guichet, A.; Olichon, A.; Chevrollier, A.; Viala, F.; Miot, S.; Ayuso, C.; Odent, S.; Arrouet, C.; Verny, C.; et al. OPA1 R445H Mutation in Optic Atrophy Associated with Sensorineural Deafness. Ann. Neurol. 2005, 58, 958–963. [Google Scholar] [CrossRef]
- Olichon, A.; Baricault, L.; Gas, N.; Guillou, E.; Valette, A.; Belenguer, P.; Lenaers, G. Loss of OPA1 Perturbates the Mitochondrial Inner Membrane Structure and Integrity, Leading to Cytochrome c Release and Apoptosis. J. Biol. Chem. 2003, 278, 7743–7746. [Google Scholar] [CrossRef] [Green Version]
- Patten, D.A.; Wong, J.; Khacho, M.; Soubannier, V.; Mailloux, R.J.; Pilon-Larose, K.; MacLaurin, J.G.; Park, D.S.; McBride, H.M.; Trinkle-Mulcahy, L.; et al. OPA1-Dependent Cristae Modulation Is Essential for Cellular Adaptation to Metabolic Demand. EMBO J. 2014, 33, 2676–2691. [Google Scholar] [CrossRef] [Green Version]
- Frezza, C.; Cipolat, S.; Martins de Brito, O.; Micaroni, M.; Beznoussenko, G.V.; Rudka, T.; Bartoli, D.; Polishuck, R.S.; Danial, N.N.; De Strooper, B.; et al. OPA1 Controls Apoptotic Cristae Remodeling Independently from Mitochondrial Fusion. Cell 2006, 126, 177–189. [Google Scholar] [CrossRef] [Green Version]
- Olichon, A.; Landes, T.; Arnauné-Pelloquin, L.; Emorine, L.J.; Mils, V.; Guichet, A.; Delettre, C.; Hamel, C.; Amati-Bonneau, P.; Bonneau, D.; et al. Effects of OPA1 Mutations on Mitochondrial Morphology and Apoptosis: Relevance to ADOA Pathogenesis. J. Cell. Physiol. 2007, 211, 423–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amati-Bonneau, P.; Milea, D.; Bonneau, D.; Chevrollier, A.; Ferré, M.; Guillet, V.; Gueguen, N.; Loiseau, D.; de Crescenzo, M.-A.P.; Verny, C.; et al. OPA1-Associated Disorders: Phenotypes and Pathophysiology. Int. J. Biochem. Cell Biol. 2009, 41, 1855–1865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu-Wai-Man, P.; Griffiths, P.G.; Gorman, G.S.; Lourenco, C.M.; Wright, A.F.; Auer-Grumbach, M.; Toscano, A.; Musumeci, O.; Valentino, M.L.; Caporali, L.; et al. Multi-System Neurological Disease Is Common in Patients with OPA1 Mutations. Brain 2010, 133, 771–786. [Google Scholar] [CrossRef] [PubMed]
- Leruez, S.; Milea, D.; Defoort-Dhellemmes, S.; Colin, E.; Crochet, M.; Procaccio, V.; Ferré, M.; Lamblin, J.; Drouin, V.; Vincent-Delorme, C.; et al. Sensorineural Hearing Loss in OPA1-Linked Disorders. Brain 2013, 136, e236. [Google Scholar] [CrossRef]
- Santarelli, R.; Rossi, R.; Scimemi, P.; Cama, E.; Valentino, M.L.; La Morgia, C.; Caporali, L.; Liguori, R.; Magnavita, V.; Monteleone, A.; et al. OPA1-Related Auditory Neuropathy: Site of Lesion and Outcome of Cochlear Implantation. Brain 2015, 138, 563–576. [Google Scholar] [CrossRef] [Green Version]
- Bette, S.; Zimmermann, U.; Wissinger, B.; Knipper, M. OPA1, the Disease Gene for Optic Atrophy Type Kjer, Is Expressed in the Inner Ear. Histochem. Cell Biol. 2007, 128, 421–430. [Google Scholar] [CrossRef]
- Huang, T.; Santarelli, R.; Starr, A. Mutation of OPA1 Gene Causes Deafness by Affecting Function of Auditory Nerve Terminals. Brain Res. 2009, 1300, 97–104. [Google Scholar] [CrossRef] [Green Version]
- Lin, P.H.; Wu, H.P.; Wu, C.M.; Chiang, Y.T.; Hsu, J.S.; Tsai, C.Y.; Wang, H.; Tseng, L.H.; Chen, P.Y.; Yang, T.H.; et al. Cochlear Implantation Outcomes in Patients with Auditory Neuropathy Spectrum Disorder of Genetic and Non-Genetic Etiologies: A Multicenter Study. Biomedicines 2022, 28, 1523. [Google Scholar] [CrossRef]
- Maeda-Katahira, A.; Nakamura, N.; Hayashi, T.; Katagiri, S.; Shimizu, S.; Ohde, H.; Matsunaga, T.; Kaga, K.; Nakano, T.; Kameya, S.; et al. Autosomal dominant optic atrophy with OPA1 gene mutations accompanied by auditory neuropathy and other systemic complications in a Japanese cohort. Mol. Vis. 2019, 25, 559–573. [Google Scholar] [PubMed]
- Foggia, M.J.; Quevedo, R.V.; Hansen, M.R. Intracochlear Fibrosis and the Foreign Body Response to Cochlear Implant Biomaterials. Laryngoscope Investig. Otolaryngol. 2019, 4, 678–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Malley, J.T.; Burgess, B.J.; Galler, D.; Nadol, J.B. Foreign Body Response to Silicone in Cochlear Implant Electrodes in the Human. Otol. Neurotol. 2017, 38, 970–977. [Google Scholar] [CrossRef] [PubMed]
- Nadol, J.B., Jr.; O’Malley, J.T.; Burgess, B.J.; Galler, D. Cellular Immunologic Responses to Cochlear Implantation in the Human. Hear. Res. 2014, 318, 11–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishiyama, A.; Doherty, J.; Ishiyama, G.; Quesnel, A.M.; Lopez, I.; Linthicum, F.H. Post Hybrid Cochlear Implant Hearing Loss and Endolymphatic Hydrops. Otol. Neurotol. 2016, 37, 1516–1521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehnhardt, E. Intracochlear placement of cochlear implant electrodes in soft surgery technique. HNO 1993, 41, 356–359. [Google Scholar]
- Cohen, N.L. Cochlear Implant Soft Surgery: Fact or Fantasy? Otolaryngol. Head Neck Surg. 1997, 117, 214–216. [Google Scholar] [CrossRef]
- Lenarz, T.; James, C.; Cuda, D.; Fitzgerald O’Connor, A.; Frachet, B.; Frijns, J.H.M.; Klenzner, T.; Laszig, R.; Manrique, M.; Marx, M.; et al. European Multi-Centre Study of the Nucleus Hybrid L24 Cochlear Implant. Int. J. Audiol. 2013, 52, 838–848. [Google Scholar] [CrossRef]
- Carlson, M.L. Cochlear Implantation in Adults. N. Engl. J. Med. 2020, 382, 1531–1542. [Google Scholar] [CrossRef]
- Van de Heyning, P.H.; Dazert, S.; Gavilan, J.; Lassaletta, L.; Lorens, A.; Rajan, G.P.; Skarzynski, H.; Skarzynski, P.H.; Tavora-Vieira, D.; Topsakal, V.; et al. Systematic Literature Review of Hearing Preservation Rates in Cochlear Implantation Associated with Medium- and Longer-Length Flexible Lateral Wall Electrode Arrays. Front. Surg. 2022, 9, 893839. [Google Scholar] [CrossRef] [PubMed]
- Weber, S.; Gavaghan, K.; Wimmer, W.; Williamson, T.; Gerber, N.; Anso, J.; Bell, B.; Feldmann, A.; Rathgeb, C.; Matulic, M.; et al. Instrument Flight to the Inner Ear. Sci. Robot. 2017, 2, eaal4916. [Google Scholar] [CrossRef] [PubMed]
- Labadie, R.F.; Balachandran, R.; Noble, J.H.; Blachon, G.S.; Mitchell, J.E.; Reda, F.A.; Dawant, B.M.; Fitzpatrick, J.M. Minimally Invasive Image-Guided Cochlear Implantation Surgery: First Report of Clinical Implementation. Laryngoscope 2014, 124, 1915–1922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Topsakal, V.; Heuninck, E.; Matulic, M.; Tekin, A.M.; Mertens, G.; Van Rompaey, V.; Galeazzi, P.; Zoka-Assadi, M.; van de Heyning, P. First Study in Men Evaluation of a Surgical Robotic Tool Providing Autonomous Inner Ear Access for Cochlear Implantation. Front. Neurol. 2022, 13, 804507. [Google Scholar] [CrossRef]
- Topsakal, V.; Matulic, M.; Assadi, M.Z.; Mertens, G.; Van Rompaey, V.; Van de Heyning, P. Comparison of the Surgical Techniques and Robotic Techniques for Cochlear Implantation in Terms of the Trajectories Toward the Inner Ear. J. Int. Adv. Otol. 2020, 16, 3–7. [Google Scholar] [CrossRef]
- Tekin, A.M.; Matulic, M.; Wuyts, W.; Assadi, M.Z.; Mertens, G.; van Rompaey, V.; Li, Y.; van de Heyning, P.; Topsakal, V. A New Pathogenic Variant in POU3F4 Causing Deafness Due to an Incomplete Partition of the Cochlea Paved the Way for Innovative Surgery. Genes 2021, 12, 613. [Google Scholar] [CrossRef]
- Kleine Punte, A.; Van de Heyning, P. Quality standards for minimal outcome measurements in adults and children. Cochlear Implants Int. 2013, 14 (Suppl. 2), 39–42. [Google Scholar] [CrossRef]
- Wouters, J.; Damman, W.; Bosman, A.J. Vlaamse Opname Van Woordenlijsten Voor Spraakaudiometrie. Logop. Inf. Vlaam. Ver. Voor Logop. 1994, 7, 28–34. [Google Scholar]
- van Wieringen, A.; Wouters, J. LIST and LINT: Sentences and numbers for quantifying speech understanding in severely impaired listeners for Flanders and the Netherlands. Int. J. Audiol. 2008, 47, 348–355. [Google Scholar] [CrossRef]
- Richards, S.; on behalf of the ACMG Laboratory Quality Assurance Committee; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [Green Version]
- Mistrík, P.; Jolly, C.; Sieber, D.; Hochmair, I. Challenging Aspects of Contemporary Cochlear Implant Electrode Array Design. World J. Otorhinolaryngol. Head Neck Surg. 2017, 3, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Wahlqvist, M.; Möller, C.; Möller, K.; Danermark, B. Physical and Psychological Health in Persons with Deafblindness That Is due to Usher Syndrome Type II. J. Vis. Impair. Blind. 2013, 107, 207–220. [Google Scholar] [CrossRef]
- Smith, R.J.; Berlin, C.I.; Hejtmancik, J.F.; Keats, B.J.; Kimberling, W.J.; Lewis, R.A.; Möller, C.G.; Pelias, M.Z.; Tranebjaerg, L. Clinical Diagnosis of the Usher Syndromes. Usher Syndrome Consortium. Am. J. Med. Genet. 1994, 50, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Health Quality Ontario. Bilateral Cochlear Implantation: A Health Technology Assessment. Ont. Health Technol. Assess. Ser. 2018, 18, 1–139. [Google Scholar]
- Liu, X.Z.; Angeli, S.I.; Rajput, K.; Yan, D.; Hodges, A.V.; Eshraghi, A.; Telischi, F.F.; Balkany, T.J. Cochlear Implantation in Individuals with Usher Type 1 Syndrome. Int. J. Pediatr. Otorhinolaryngol. 2008, 72, 841–847. [Google Scholar] [CrossRef]
- Henricson, C.; Wass, M.; Lidestam, B.; Möller, C.; Lyxell, B. Cognitive Skills in Children with Usher Syndrome Type 1 and Cochlear Implants. Int. J. Pediatr. Otorhinolaryngol. 2012, 76, 1449–1457. [Google Scholar] [CrossRef]
- Damen, G.W.J.A.; Pennings, R.J.E.; Snik, A.F.M.; Mylanus, E.A.M. Quality of Life and Cochlear Implantation in Usher Syndrome Type I. Laryngoscope 2006, 116, 723–728. [Google Scholar] [CrossRef]
- Hartel, B.P.; van Nierop, J.W.I.; Huinck, W.J.; Rotteveel, L.J.C.; Mylanus, E.A.M.; Snik, A.F.; Kunst, H.P.M.; Pennings, R.J.E. Cochlear Implantation in Patients With Usher Syndrome Type IIa Increases Performance and Quality of Life. Otol. Neurotol. 2017, 38, e120–e127. [Google Scholar] [CrossRef]
- Jatana, K.R.; Thomas, D.; Weber, L.; Mets, M.B.; Silverman, J.B.; Young, N.M. Usher Syndrome: Characteristics and Outcomes of Pediatric Cochlear Implant Recipients. Otol. Neurotol. 2013, 34, 484–489. [Google Scholar] [CrossRef]
- Santarelli, R. Information from Cochlear Potentials and Genetic Mutations Helps Localize the Lesion Site in Auditory Neuropathy. Genome Med. 2010, 2, 91. [Google Scholar] [CrossRef] [Green Version]
- Shearer, A.E.; Hansen, M.R. Auditory Synaptopathy, Auditory Neuropathy, and Cochlear Implantation. Laryngoscope Investig. Otolaryngol. 2019, 4, 429–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez-Ballesteros, M.; del Castillo, F.J.; Martín, Y.; Moreno-Pelayo, M.A.; Morera, C.; Prieto, F.; Marco, J.; Morant, A.; Gallo-Terán, J.; Morales-Angulo, C.; et al. Auditory Neuropathy in Patients Carrying Mutations in the Otoferlin Gene (OTOF). Hum. Mutat. 2003, 22, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Anderson, P.; Schaefer, S.; Henderson, L.; Bruce, I.A. Cochlear Implantation in Children with Auditory Neuropathy: Lessons from Brown-Vialetto-Van Laere Syndrome. Cochlear Implants Int. 2019, 20, 31–38. [Google Scholar] [CrossRef]
- Miyamoto, R.T.; Kirk, K.I.; Renshaw, J.; Hussain, D. Cochlear Implantation in Auditory Neuropathy. Laryngoscope 1999, 109, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Brookes, J.T.; Kanis, A.B.; Tan, L.Y.; Tranebjaerg, L.; Vore, A.; Smith, R.J.H. Cochlear Implantation in Deafness-Dystonia-Optic Neuronopathy (DDON) Syndrome. Int. J. Pediatr. Otorhinolaryngol. 2008, 72, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, C.R.; Henslee, A.M.; Claussen, A.; Hansen, M.R. Evaluation of Insertion Forces and Cochlea Trauma Following Robotics-Assisted Cochlear Implant Electrode Array Insertion. Otol. Neurotol. 2020, 41, 631–638. [Google Scholar] [CrossRef] [PubMed]
- Wimmer, W.; Venail, F.; Williamson, T.; Akkari, M.; Gerber, N.; Weber, S.; Caversaccio, M.; Uziel, A.; Bell, B. Semiautomatic Cochleostomy Target and Insertion Trajectory Planning for Minimally Invasive Cochlear Implantation. Biomed. Res. Int. 2014, 2014, 596498. [Google Scholar] [CrossRef] [Green Version]
- Bom Braga, G.O.; Schneider, D.; Muller, F.; Hermann, J.; Weber, S.; Caversaccio, M. Feasibility of Pediatric Robotic Cochlear Implantation in Phantoms. Otol. Neurotol. 2020, 41, e192–e200. [Google Scholar] [CrossRef]
- Carelli, V.; Sabatelli, M.; Carrozzo, R.; Rizza, T.; Schimpf, S.; Wissinger, B.; Zanna, C.; Rugolo, M.; La Morgia, C.; Caporali, L.; et al. ‘Behr syndrome’ with OPA1 compound heterozygote mutations. Brain J. Neurol. 2015, 138, e321. [Google Scholar] [CrossRef] [Green Version]
- Bonifert, T.; Karle, K.N.; Tonagel, F.; Batra, M.; Wilhelm, C.; Theurer, Y.; Schoenfeld, C.; Kluba, T.; Kamenisch, Y.; Carelli, V.; et al. Pure and syndromic optic atrophy explained by deep intronic OPA1 mutations and an intralocus modifier. Brain 2014, 137, 2164–2177. [Google Scholar] [CrossRef] [Green Version]
- Schaaf, C.P.; Blazo, M.; Lewis, R.A.; Tonini, R.E.; Takei, H.; Wang, J.; Wong, L.J.; Scaglia, F. Early-onset severe neuromuscular phenotype associated with compound heterozygosity for OPA1 mutations. Mol. Genet. Metab. 2011, 103, 383–387. [Google Scholar] [CrossRef]
- Bonneau, D.; Colin, E.; Oca, F.; Ferré, M.; Chevrollier, A.; Guéguen, N.; Desquiret-Dumas, V.; N’Guyen, S.; Barth, M.; Zanlonghi, X.; et al. Early-onset Behr syndrome due to compound heterozygous mutations in OPA1. Brain 2014, 137, e301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nasca, A.; Rizza, T.; Doimo, M.; Legati, A.; Ciolfi, A.; Diodato, D.; Calderan, C.; Carrara, G.; Lamantea, E.; Aiello, C.; et al. Not only dominant, not only optic atrophy: Expanding the clinical spectrum associated with OPA1 mutations. Orphanet J. Rare Dis. 2017, 12, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weisschuh, N.; Schimpf-Linzenbold, S.; Mazzola, P.; Kieninger, S.; Xiao, T.; Kellner, U.; Neuhann, T.; Kelbsch, C.; Tonagel, F.; Wilhelm, H.; et al. Mutation spectrum of the OPA1 gene in a large cohort of patients with suspected dominant optic atrophy: Identification and classification of 48 novel variants. PLoS ONE 2021, 16, e0253987. [Google Scholar] [CrossRef] [PubMed]
- Thiselton, D.L.; Alexander, C.; Taanman, J.W.; Brooks, S.; Rosenberg, T.; Eiberg, H.; Andreasson, S.; Van Regemorter, N.; Munier, F.L.; Moore, A.T.; et al. A comprehensive survey of mutations in the OPA1 gene in patients with autosomal dominant optic atrophy. Investig. Ophthalmol. Vis. Sci. 2002, 43, 1715–1724. [Google Scholar]
- Toomes, C.; Marchbank, N.J.; Mackey, D.A.; Craig, J.E.; Newbury-Ecob, R.A.; Bennett, C.P.; Vize, C.J.; Desai, S.P.; Black, G.C.; Patel, N.; et al. Spectrum, frequency and penetrance of OPA1 mutations in dominant optic atrophy. Hum. Mol. Genet. 2001, 10, 1369–1378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohn, A.C.; Toomes, C.; Potter, C.; Towns, K.V.; Hewitt, A.W.; Inglehearn, C.F.; Craig, J.E.; Mackey, D.A. Autosomal dominant optic atrophy: Penetrance and expressivity in patients with OPA1 mutations. Am. J. Ophthalmol. 2007, 143, 656–662. [Google Scholar] [CrossRef] [PubMed]
- Fuhrmann, N.; Schimpf, S.; Kamenisch, Y.; Leo-Kottler, B.; Alexander, C.; Auburger, G.; Zrenner, E.; Wissinger, B.; Alavi, M.V. Solving a 50 year mystery of a missing OPA1 mutation: More insights from the first family diagnosed with autosomal dominant optic atrophy. Mol. Neurodegener. 2010, 5, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, T.; Ye, B.; Chen, K.; Zhang, A.; Guo, D.; Pan, Y.; Ding, R.; Hu, H.; Sun, X.; Xiang, M. Impacts of impaired mitochondrial dynamics in hearing loss: Potential therapeutic targets. Front. Neurosci. 2022, 16, 998507. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
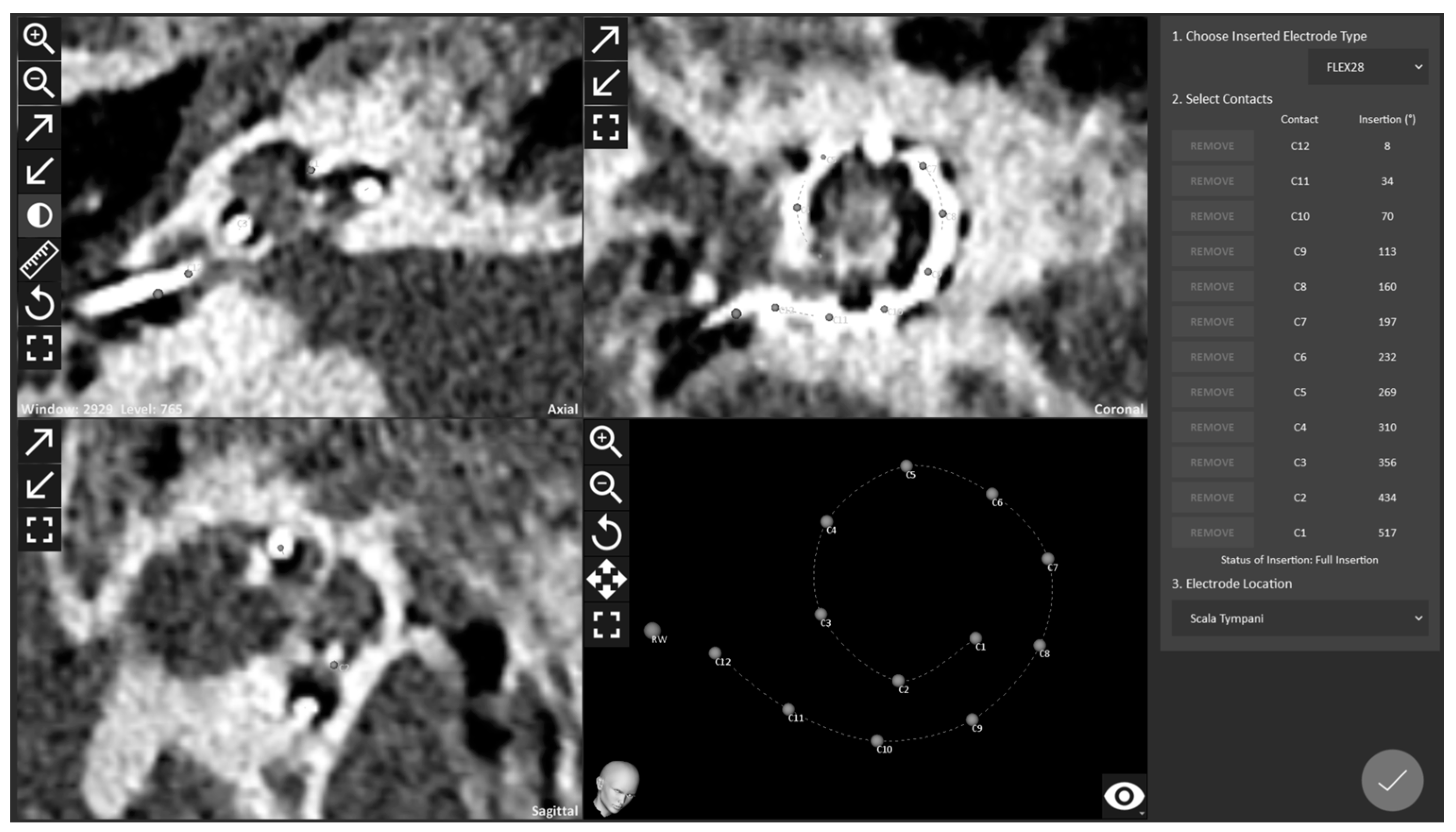{kind=link}
{kind=link}
| Nucleotide Change | Amino Acid Change | Feature of Deafness | Cochlear Implantation (CI) | Improvements in Auditory and Speech Performance after CI | Location | Year | References |
|---|---|---|---|---|---|---|---|
| c.1316 G>T | p.G439V | SNHL | Unilateral | No | Italy | 2015 | [21] |
| c.1316 G>T | p.G439V | SNHL | Unilateral | Yes | Italy | 2015 | [21] |
| c.869 G>A | p.R290Q | SNHL | Unilateral | Yes | Italy | 2015 | [21] |
| c.1334 G>A | p.R445H | SNHL | Unilateral | Yes | Italy | 2015 | [21] |
| c.1334 G>A | p.R445H | SNHL | Unilateral | Yes | Italy | 2015 | [21] |
| c.893 G>A | p.S298N | SNHL | Unilateral | Yes | Italy | 2015 | [21] |
| c.1334 G>A | p.R445H | SNHL | Unilateral | Yes | USA | 2015 | [21,23] |
| c.1334 G>A | p.R445H | SNHL | Unilateral | Yes | USA | 2015 | [21,23] |
| c.892A>C | p.Ser298Arg | SNHL | Unilateral | Yes | Japan | 2019 | [25] |
| c.1334G>A | p.Arg445His | SNHL | Unilateral | Yes | Japan | 2019 | [25] |
| c.1414T>C | p.Cys472Arg | SNHL | Bilateral | Yes | Taiwan | 2022 | [24] |
| c.1499G>A | p.(Arg500His) | SNHL | Bilateral | Yes | Belgium | 2022 | Present Study |
| Implanted Ear | Type of Surgery | Age at Implantation (Years) | Inactive Electrodes | PTA0.5; 1; 2 and 4 kHz (in dB HL) | Speech in Quiet at 65 dB SPL (% Correct) | Speech in Noise (dB SNR) | |||
|---|---|---|---|---|---|---|---|---|---|
| Pre-op Unaided | Post-op CI Ear (2 Years) | Pre-op Unaided | Post-op CI Ear (2 Years) | Pre-op Unaided | Post-op CI Ear (2 Years) | ||||
| Left | Conventional | 36 | / | 105 | 40 | 0 | 91 | >20 | +2 |
| Right | RACIS | 38 | / | 95 | 30 | 0 | 88 | > 20 | +0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tekin, A.M.; Baelen, H.; Heuninck, E.; Bayazıt, Y.A.; Mertens, G.; Rompaey, V.v.; Heyning, P.v.d.; Topsakal, V. Evaluation of a Less Invasive Cochlear Implant Surgery in OPA1 Mutations Provoking Deafblindness. Genes 2023, 14, 627. https://doi.org/10.3390/genes14030627
Tekin AM, Baelen H, Heuninck E, Bayazıt YA, Mertens G, Rompaey Vv, Heyning Pvd, Topsakal V. Evaluation of a Less Invasive Cochlear Implant Surgery in OPA1 Mutations Provoking Deafblindness. Genes. 2023; 14(3):627. https://doi.org/10.3390/genes14030627
Chicago/Turabian StyleTekin, Ahmet M., Hermine Baelen, Emilie Heuninck, Yıldırım A. Bayazıt, Griet Mertens, Vincent van Rompaey, Paul van de Heyning, and Vedat Topsakal. 2023. "Evaluation of a Less Invasive Cochlear Implant Surgery in OPA1 Mutations Provoking Deafblindness" Genes 14, no. 3: 627. https://doi.org/10.3390/genes14030627