
Accumulation of STR-Loci Aberrations in Subclones of Jurkat Cell Line as a Model of Tumor Clonal Evolution

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
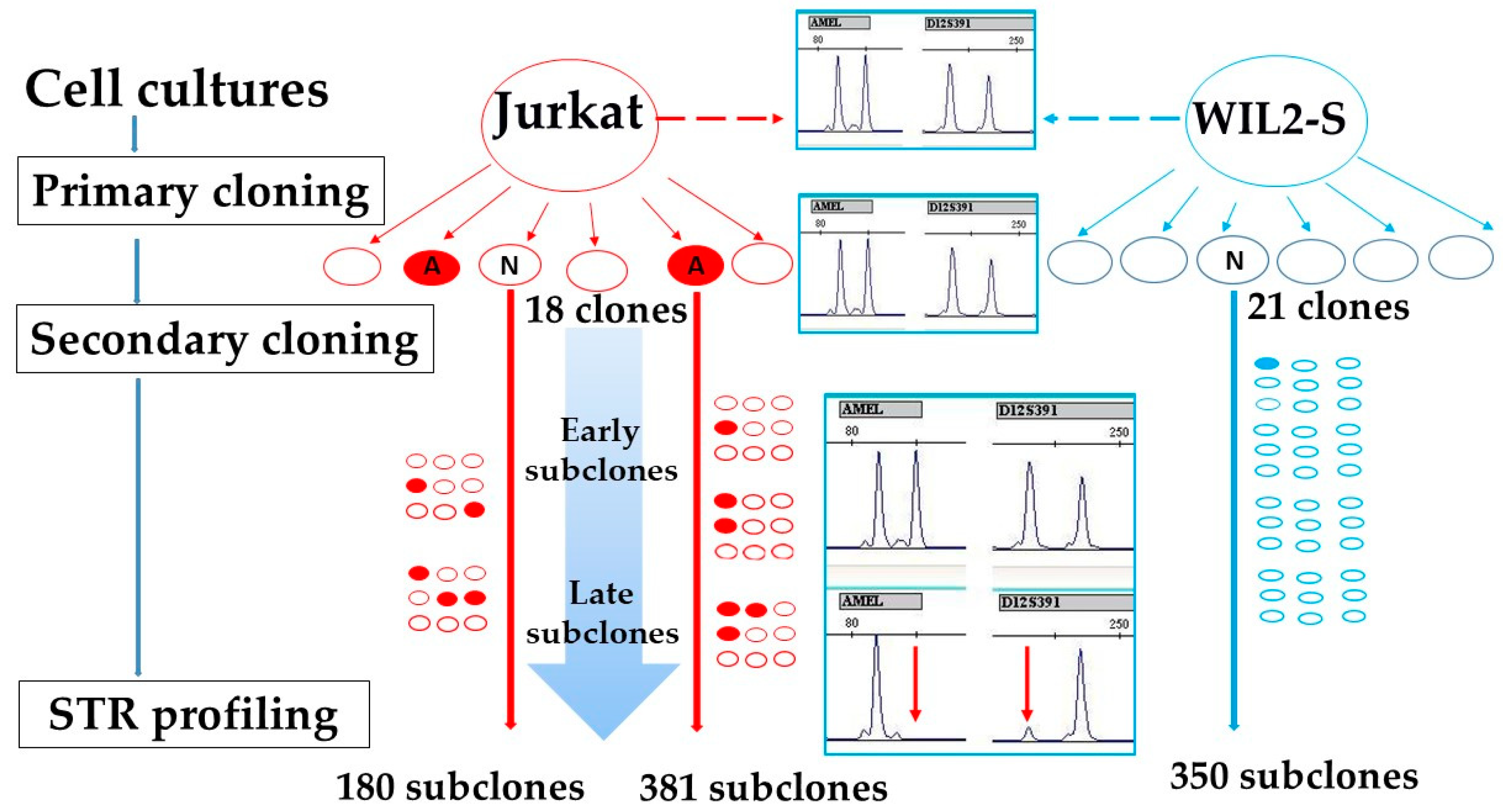
2.1. Cell Cultures Cloning by Limiting Dilutions
2.2. DNA Isolation
2.3. STR Profiling
2.4. Statistical Analysis
3. Results
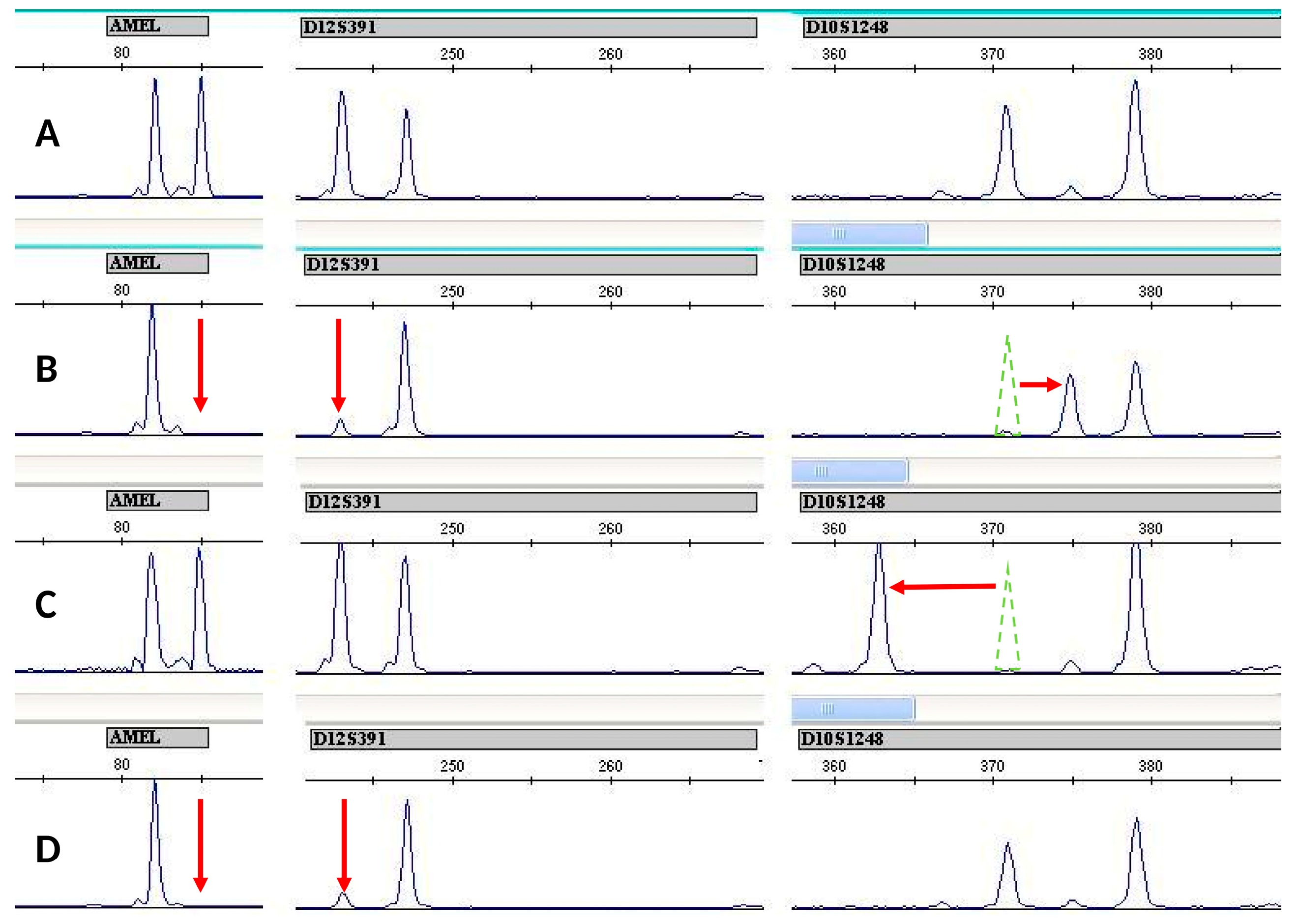
3.1. Cell Lines Verification by STR Profiling
3.2. WIL2-S and Jurkat Primary Clones Analysis
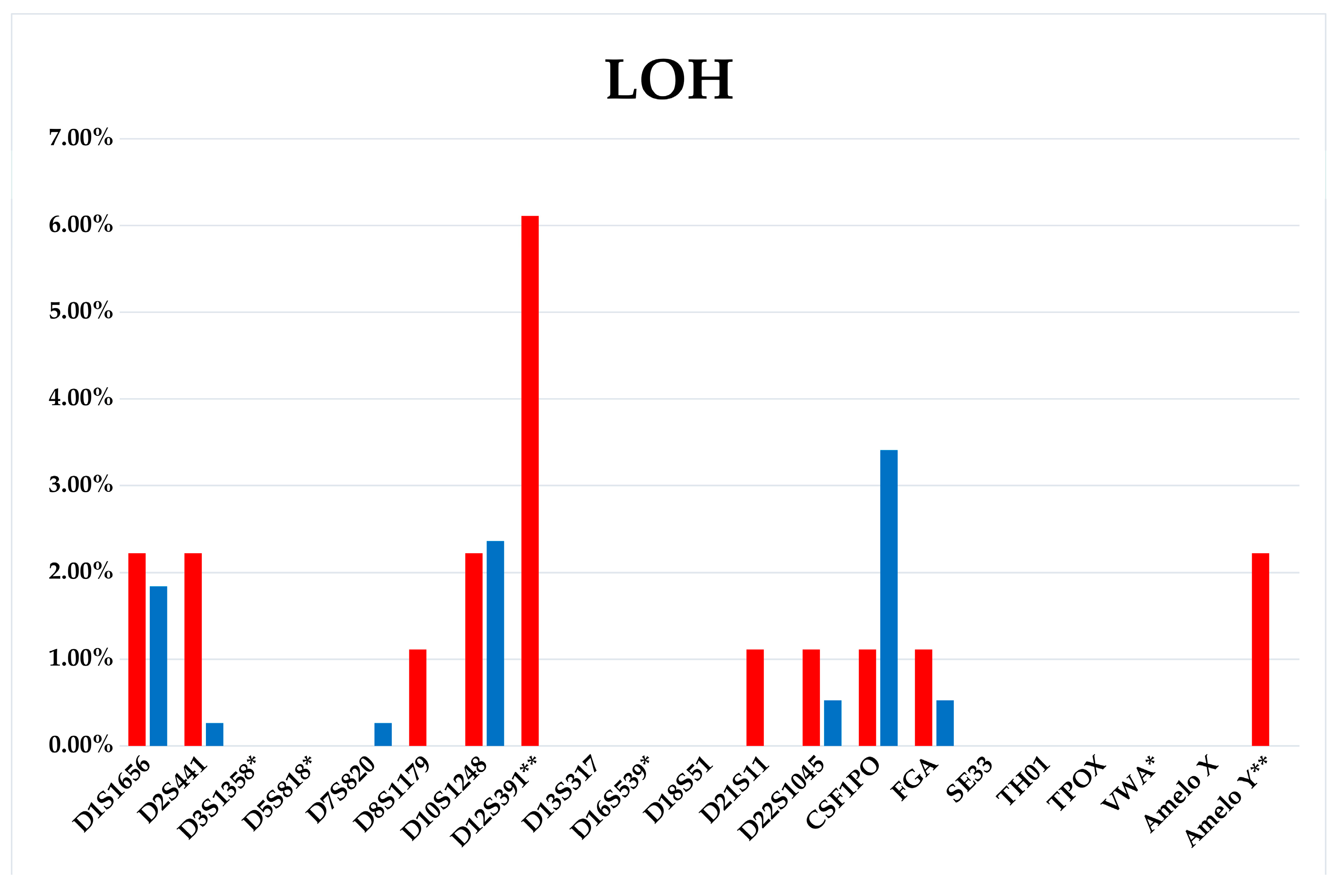
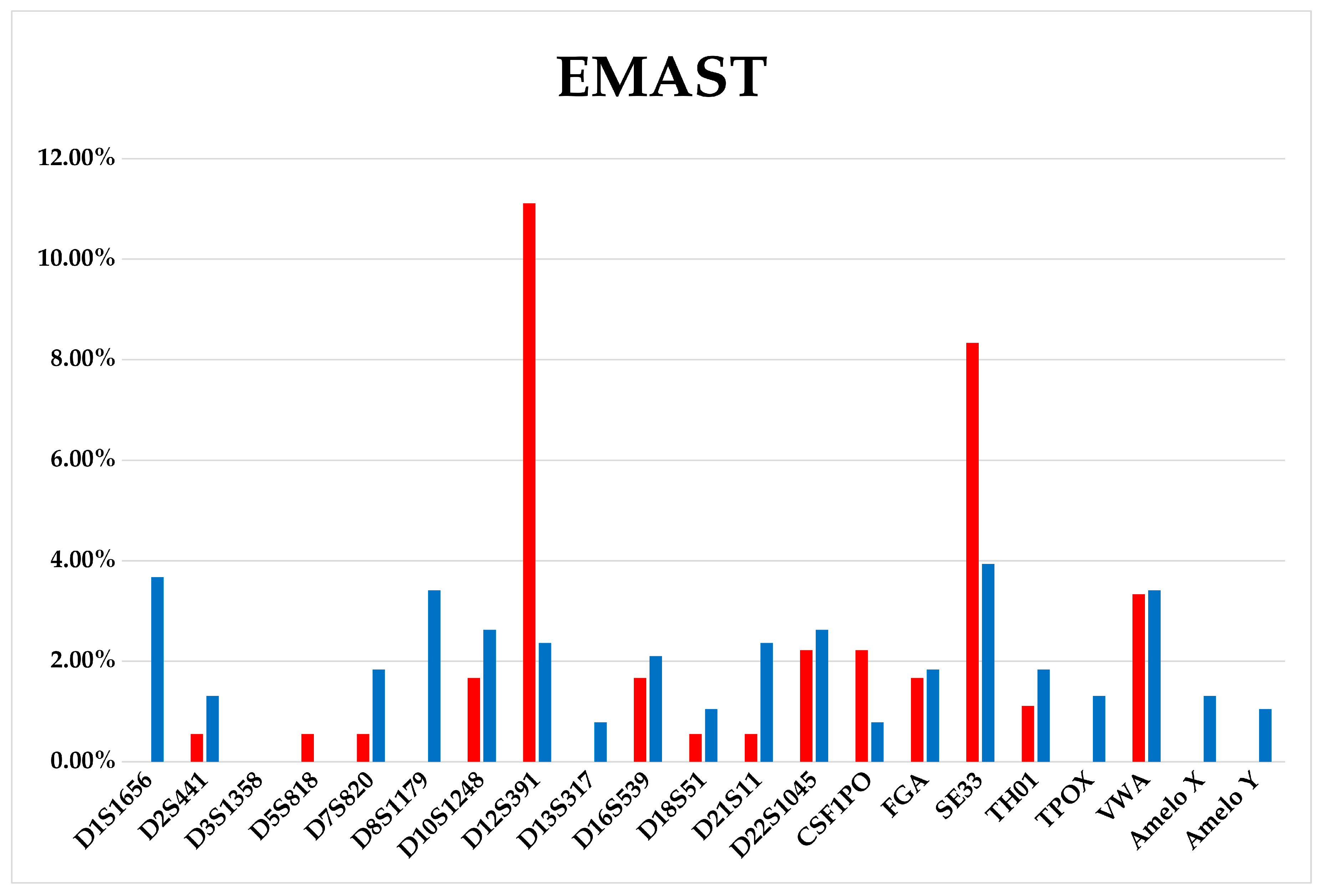
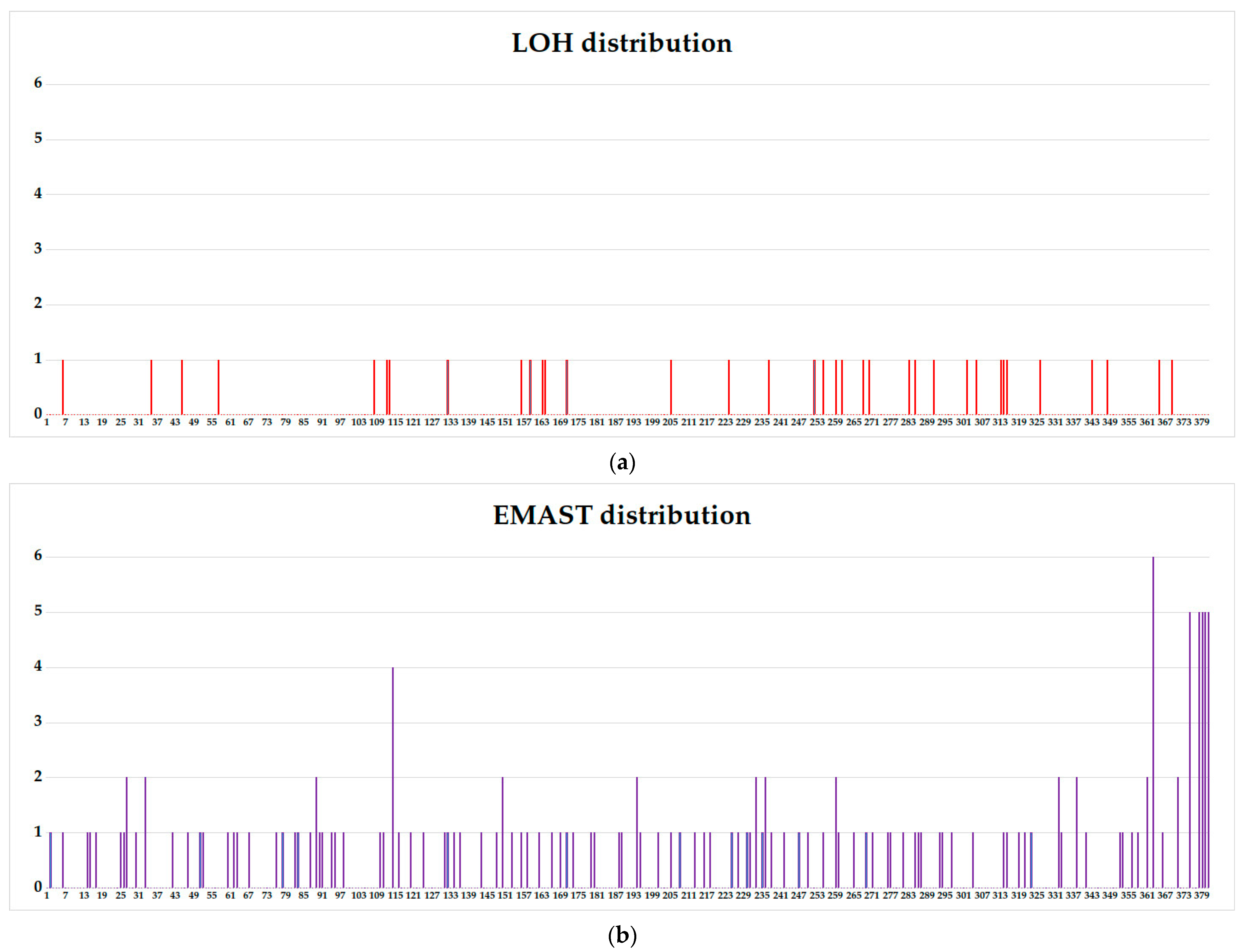
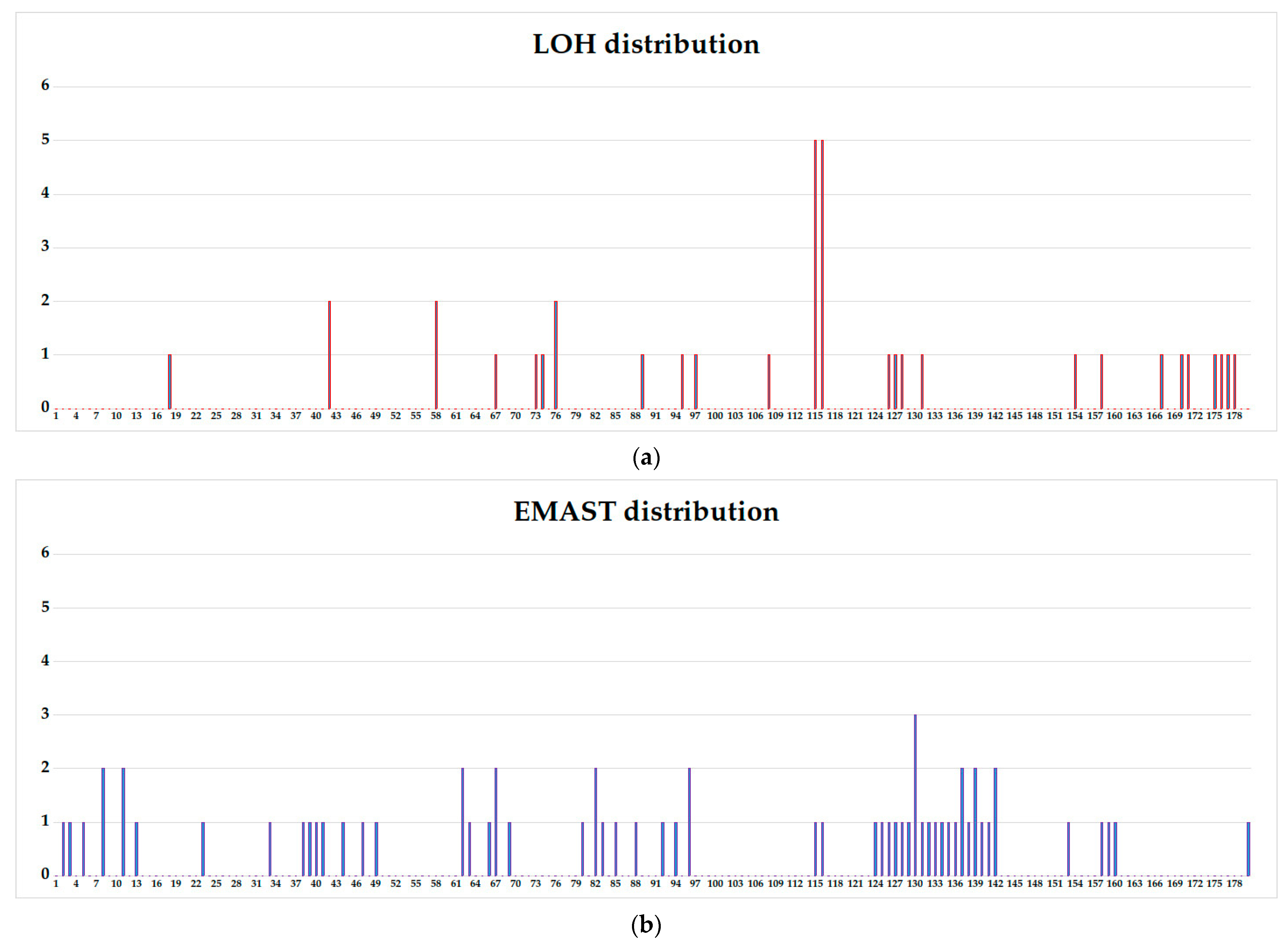
3.3. WIL2-S and Jurkat Subclones Analysis
3.4. Comparison of Aberrations Frequencies in Fast-Growing, Intermediate, and Slow-Growing Jurkat Subclones
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef] [Green Version]
- Rampias, T. Exploring the Eco-Evolutionary Dynamics of Tumor Subclones. Cancers 2020, 12, 3436. [Google Scholar] [CrossRef]
- Phillips, D.H. Mutational spectra and mutational signatures: Insights into cancer aetiology and mechanisms of DNA damage and repair. DNA Repair (Amst) 2018, 71, 6–11. [Google Scholar] [CrossRef]
- Freeman, S.D.; Hourigan, C.S. MRD evaluation of AML in clinical practice: Are we there yet? Hematology Am Soc Hematol Educ Program 2019, 6, 557–569. [Google Scholar] [CrossRef]
- Kövy, P.; Őrfi, Z.; Bors, A.; Kozma, A.; Gopcsa, L.; Dolgos, J.; Lovas, N.; Harasztdombi, J.; Lakatos, V.; Király, Á.; et al. Nucleophosmin1 and isocitrate dehydrogenase 1 and 2 as measurable residual disease markers in acute myeloid leukemia. PLoS ONE 2021, 16, e0253386. [Google Scholar] [CrossRef] [PubMed]
- Rapaport, F.; Neelamraju, Y.; Baslan, T.; Hassane, D.; Gruszczynska, A.; Robert de Massy, M.; Farnoud, N.; Haddox, S.; Lee, T.; Medina-Martinez, J.; et al. Genomic and evolutionary portraits of disease relapse in acute myeloid leukemia. Leukemia 2021, 35, 2688–2692. [Google Scholar] [CrossRef] [PubMed]
- Worsley, C.M.; Mayne, E.S.; Veale, R.B. Clone wars: The evolution of therapeutic resistance in cancer. Evol. Med. Public Health 2016, 2016, 180–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foo, J.; Michor, F. Evolution of acquired resistance to anti-cancer therapy. J. Theor. Biol. 2014, 355, 10–20. [Google Scholar] [CrossRef] [Green Version]
- Marusyk, A.; Janiszewska, M.; Polyak, K. Intratumor Heterogeneity: The Rosetta Stone of Therapy Resistance. Cancer Cell 2020, 37, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Nicoś, M.; Krawczyk, P.; Crosetto, N.; Milanowski, J. The Role of Intratumor Heterogeneity in the Response of Metastatic Non-Small Cell Lung Cancer to Immune Checkpoint Inhibitors. Front. Oncol. 2020, 10, 569202. [Google Scholar] [CrossRef]
- Waanders, E.; Gu, Z.; Dobson, S.M.; Antić, Ž.; Crawford, J.C.; Ma, X.; Edmonson, M.N.; Payne-Turner, D.; van de Vorst, M.; Jongmans, M.C.J.; et al. Mutational landscape and patterns of clonal evolution in relapsed pediatric acute lymphoblastic leukemia. Blood Cancer Discov. 2020, 1, 96–111. [Google Scholar] [CrossRef]
- Spinella, J.F.; Richer, C.; Cassart, P.; Ouimet, M.; Healy, J.; Sinnett, D. Mutational dynamics of early and late relapsed childhood ALL: Rapid clonal expansion and long-term dormancy. Blood Adv. 2018, 2, 177–188. [Google Scholar] [CrossRef] [Green Version]
- Risinskaya, N.; Mangasarova, Y.; Nikulina, E.; Kozhevnikova, Y.; Chabaeva, J.; Yushkova, A.; Magomedova, A.; Kulikov, S.; Julhakyan, H.; Kravchenko, S.; et al. STR Profiling Reveals Tumor Genome Instability in Primary Mediastinal B-Cell Lymphoma. Curr. Oncol. 2022, 29, 3449–3459. [Google Scholar] [CrossRef]
- Risinskaya, N.; Kozhevnikova, Y.; Gavrilina, O.; Chabaeva, J.; Kotova, E.; Yushkova, A.; Isinova, G.; Zarubina, K.; Obukhova, T.; Kulikov, S.; et al. Loss of Heterozygosity in the Tumor DNA of De Novo Diagnosed Patients Is Associated with Poor Outcome for B-ALL but Not for T-ALL. Genes 2022, 13, 398. [Google Scholar] [CrossRef]
- Soloveva, M.; Solovev, M.; Nikulina, E.; Risinskaya, N.; Biderman, B.; Yakutik, I.; Obukhova, T.; Mendeleeva, L. Loss of Heterozygosity in the Circulating Tumor DNA and CD138+ Bone Marrow Cells in Multiple Myeloma. Genes 2023, 14, 351. [Google Scholar] [CrossRef] [PubMed]
- Firsova, M.V.; Risinskaya, N.V.; Solovev, M.V.; Obukhova, T.N.; Kislitsyna, M.A.; Nikulina, E.E.; Yakutik, I.A.; Abramova, T.V.; Sudarikov, A.B.; Kovrigina, A.M.; et al. Multiple myeloma with extramedullary plasmacytoma: Pathogenesis and clinical case. Oncohematology 2022, 17, 67–80. (In Russian) [Google Scholar] [CrossRef]
- Liu, L.; Cao, F.; Wang, S.; Zhou, J.; Yang, G.; Wang, C. Detection of malignant B lymphocytes by PCR clonality assay using direct lysis of cerebrospinal fluid and low volume specimens. Int. J. Lab. Hematol. 2015, 37, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Khorolsky, M.; Semenova, I.; Melnikova, E.; Olefir, Y. The Use of Short Tandem Repeat Analysis for Cell Line Authentication. Bioprep. Prev. Diagn. Treat. 2019, 19, 251–260. [Google Scholar] [CrossRef]
- Nowell, P.C. The clonal evolution of tumor cell populations. Science 1976, 194, 23–28. [Google Scholar] [CrossRef]
- Fidler, I.J.; Kripke, M.L. Metastasis results from preexisting variant cells within a malignant tumor. Science 1977, 197, 893–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; Tarpey, P.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreso, A.; O’Brien, C.A.; van Galen, P.; Gan, O.I.; Notta, F.; Brown, A.M.; Ng, K.; Ma, J.; Wienholds, E.; Dunant, C.; et al. Variable clonal repopulation dynamics influence chemotherapy response in colorectal cancer. Science 2013, 339, 543–548. [Google Scholar] [CrossRef] [Green Version]
- Ding, L.; Ley, T.J.; Larson, D.E.; Miller, C.A.; Koboldt, D.C.; Welch, J.S.; Ritchey, J.K.; Young, M.A.; Lamprecht, T.; McLellan, M.D.; et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 2012, 481, 506–510. [Google Scholar] [CrossRef] [Green Version]
- Keats, J.J.; Chesi, M.; Egan, J.B.; Garbitt, V.M.; Palmer, S.E.; Braggio, E.; Van Wier, S.; Blackburn, P.R.; Baker, A.S.; Dispenzieri, A.; et al. Clonal competition with alternating dominance in multiple myeloma. Blood 2012, 120, 1067–1076. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Redmond, D.; Nie, K.; Eng, K.W.; Clozel, T.; Martin, P.; Tan, L.H.; Melnick, A.M.; Tam, W.; Elemento, O. Deep sequencing reveals clonal evolution patterns and mutation events associated with relapse in B-cell lymphomas. Genome Biol. 2014, 15, 432. [Google Scholar] [PubMed] [Green Version]
- Oshima, K.; Khiabanian, H.; da Silva-Almeida, A.C.; Tzoneva, G.; Abate, F.; Ambesi-Impiombato, A.; Sanchez-Martin, M.; Carpenter, Z.; Penson, A.; Perez-Garcia, A.; et al. Mutational landscape, clonal evolution patterns, and role of RAS mutations in relapsed acute lymphoblastic leukemia. Proc. Natl. Acad. Sci. USA 2016, 113, 11306–11311. [Google Scholar] [CrossRef] [Green Version]
- Smirnova, S.Y.; Sidorova, Y.V.; Ryzhikova, N.V.; Sychevskaya, K.A.; Parovichnikova, E.N.; Sudarikov, A.B. Evolution of Tumor Clones in Adult Acute Lymphoblastic Leukemia. Acta Nat. 2016, 8, 100–109. [Google Scholar] [CrossRef]
- Malcikova, J.; Stano-Kozubik, K.; Tichy, B.; Kantorova, B.; Pavlova, S.; Tom, N.; Radova, L.; Smardova, J.; Pardy, F.; Doubek, M.; et al. Detailed analysis of therapy-driven clonal evolution of TP53 mutations in chronic lymphocytic leukemia. Leukemia 2015, 29, 877–885. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Gao, X.; Zheng, Z.; Zhao, X.; Zhang, S.; Li, C.; Liu, G. Intratumor Heterogeneity as a Prognostic Factor in Solid Tumors: A Systematic Review and Meta-Analysis. Front. Oncol. 2021, 11, 744064. [Google Scholar] [CrossRef]
- Su, N.; Li, Z.; Yang, J.; Fu, Y.; Zhu, X.; Miao, H.; Yu, Y.; Jiang, W.; Le, J.; Qian, X.; et al. Revealing the intratumoral heterogeneity of non-DS acute megakaryoblastic leukemia in single-cell resolution. Front. Oncol. 2022, 12, 915833. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| STR Locus | WIL2-S Alleles 1 | WIL2-S Alleles 3 | Jurkat Alleles 2 | Jurkat Alleles 3 |
|---|---|---|---|---|
| Amelogenin | X,Y | X,Y | X,Y | X,Y |
| CSF1PO | 11,12 | 11,12 | 11,12 | 11,12 |
| D13S317 | 11 | 11 | 8,12 | 8,12 |
| D16S539 | 11,12 | 11,12 | 11 | 11 |
| D5S818 | 12,13 | 12,13 | 9 | 9 |
| D7S820 | 9,11 | 9,11 | 8,12 | 8,12 |
| THO1 | 8,9.3 | 8,9.3 | 6,9.3 | 6,9.3 |
| THPOX | 8,11 | 8,11 | 8,10 | 8,10 |
| vWA | 17,20 | 17,20 | 18 | 18 |
| D1S1656 | - | 14,15.3 | - | 15.3,16.3 |
| D2S441 | - | 12,15 | - | 15,16 |
| D3S1358 | - | 16 | - | 15 |
| D8S1179 | - | 10,13 | - | 13,14 |
| D10S1248 | - | 14,16 | - | 14,16 |
| D12S391 | - | 17,22 | - | 22,23 |
| D18S51 | - | 11,16 | - | 12.2,21 |
| D21S11 | - | 28 | - | 30.2,32.2 |
| D22S1045 | - | 15,16 | - | 14,17 |
| FGA | - | 20,22 | - | 20.2,21.2 |
| SE33 | - | 16,18 | - | 16,18 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Risinskaya, N.; Glinshchikova, O.; Makarik, T.; Kozhevnikova, Y.; Chabaeva, J.; Kulikov, S. Accumulation of STR-Loci Aberrations in Subclones of Jurkat Cell Line as a Model of Tumor Clonal Evolution. Genes 2023, 14, 571. https://doi.org/10.3390/genes14030571
Risinskaya N, Glinshchikova O, Makarik T, Kozhevnikova Y, Chabaeva J, Kulikov S. Accumulation of STR-Loci Aberrations in Subclones of Jurkat Cell Line as a Model of Tumor Clonal Evolution. Genes. 2023; 14(3):571. https://doi.org/10.3390/genes14030571
Chicago/Turabian StyleRisinskaya, Natalya, Olga Glinshchikova, Tatiana Makarik, Yana Kozhevnikova, Julia Chabaeva, and Sergey Kulikov. 2023. "Accumulation of STR-Loci Aberrations in Subclones of Jurkat Cell Line as a Model of Tumor Clonal Evolution" Genes 14, no. 3: 571. https://doi.org/10.3390/genes14030571