
Stratification of a Phelan–McDermid Syndrome Population Based on Their Response to Human Growth Hormone and Insulin-like Growth Factor

 , , ,
, , , 
Abstract
:1. Introduction
2. Methods
2.1. Participants
2.2. Lymphoblastoid Cell Lines (LCLs)
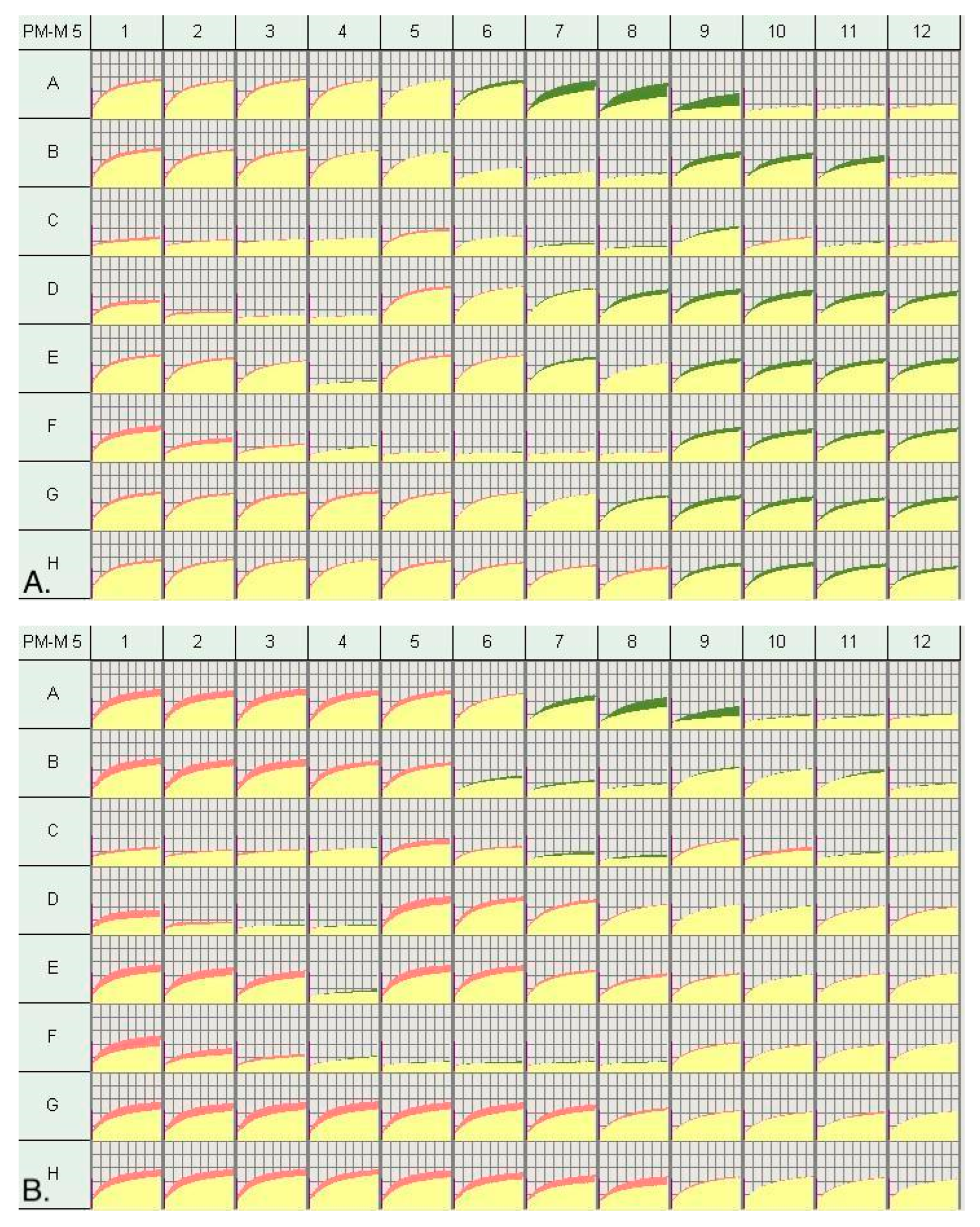
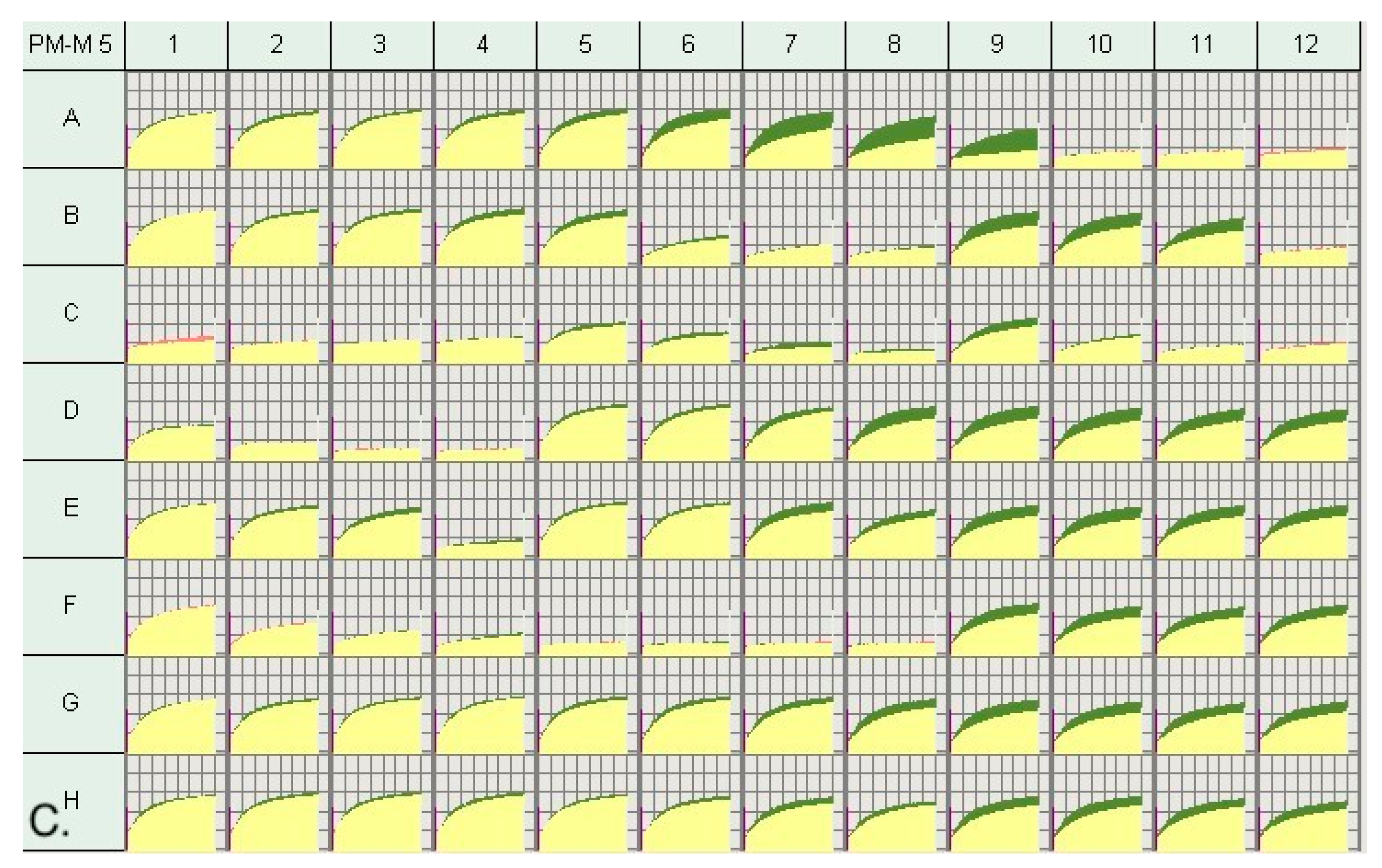
2.3. Metabolic Profiling via Biolog Phenotype Mammalian Microarrays (PM-Ms)
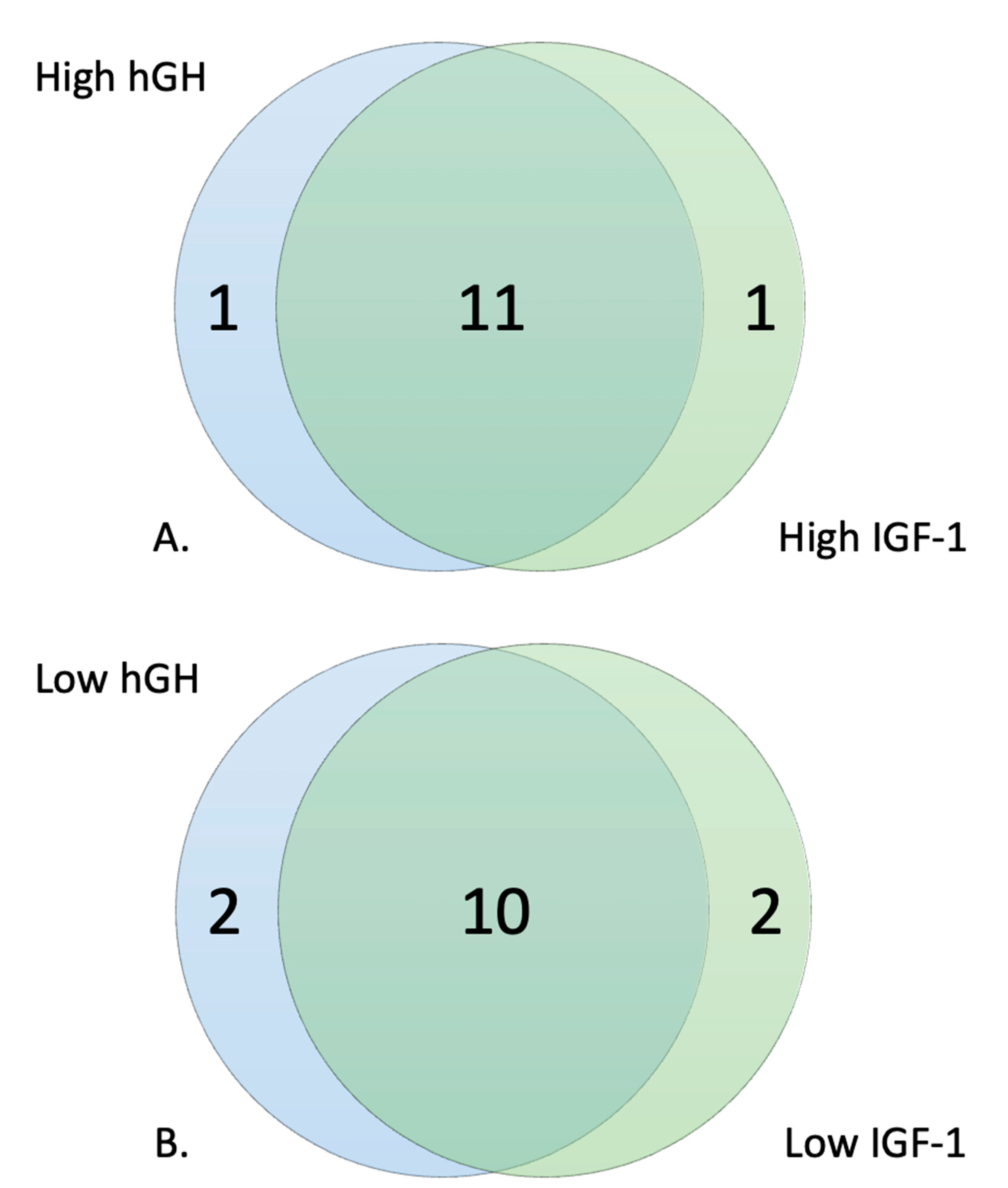
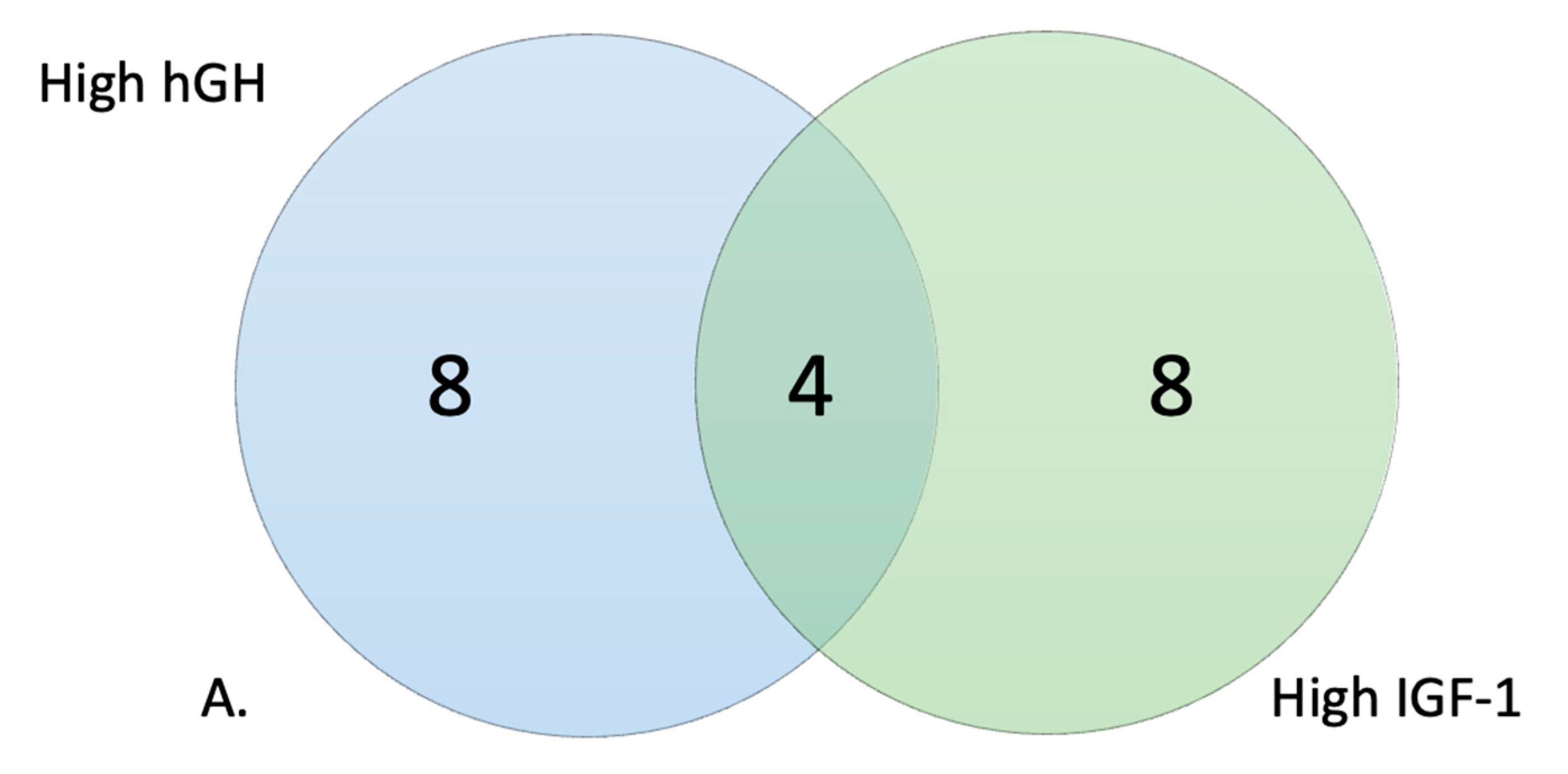
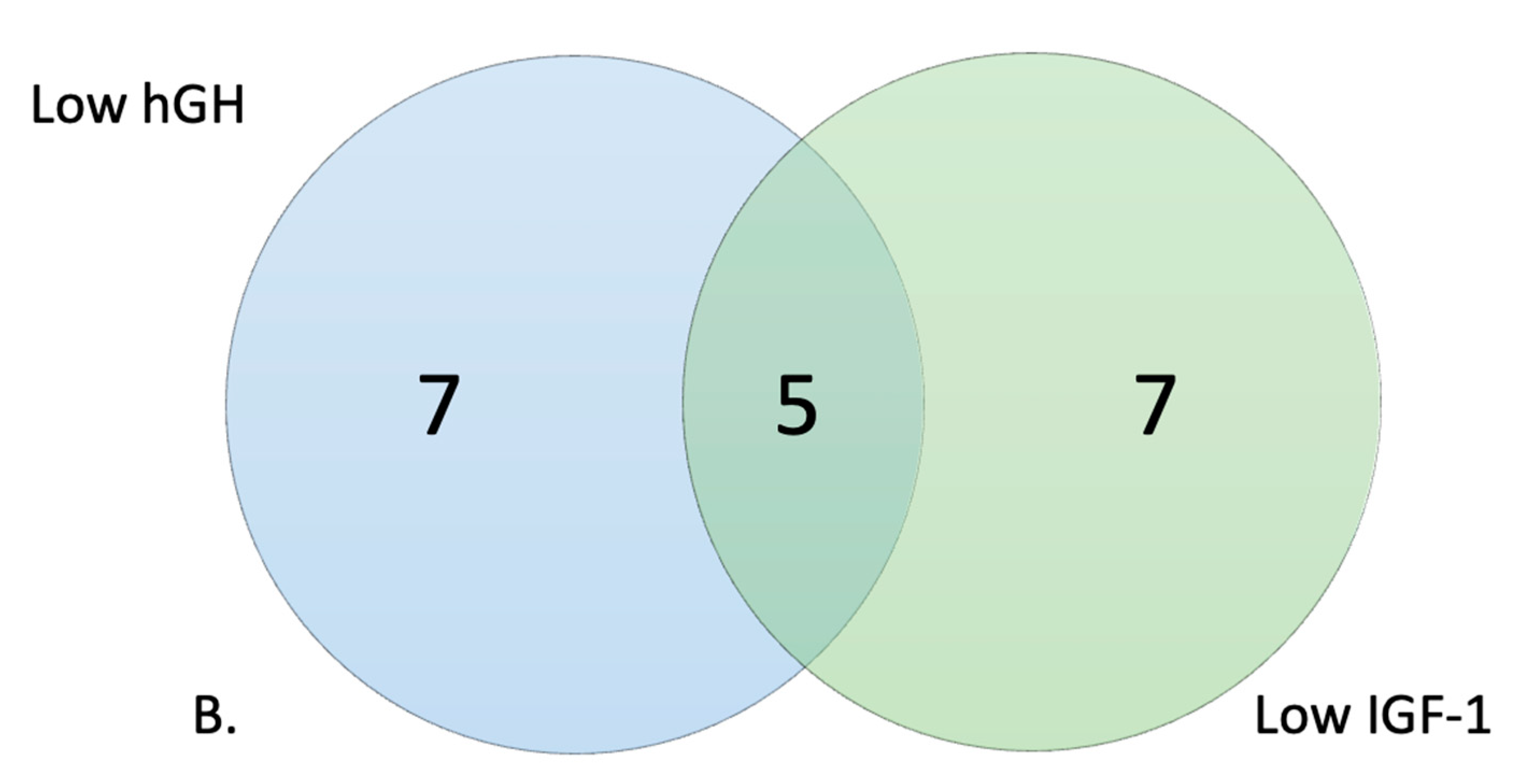
2.4. Selection of High and Low Responders
2.5. Correlation to Account for Baseline Cellular Response
3. Results
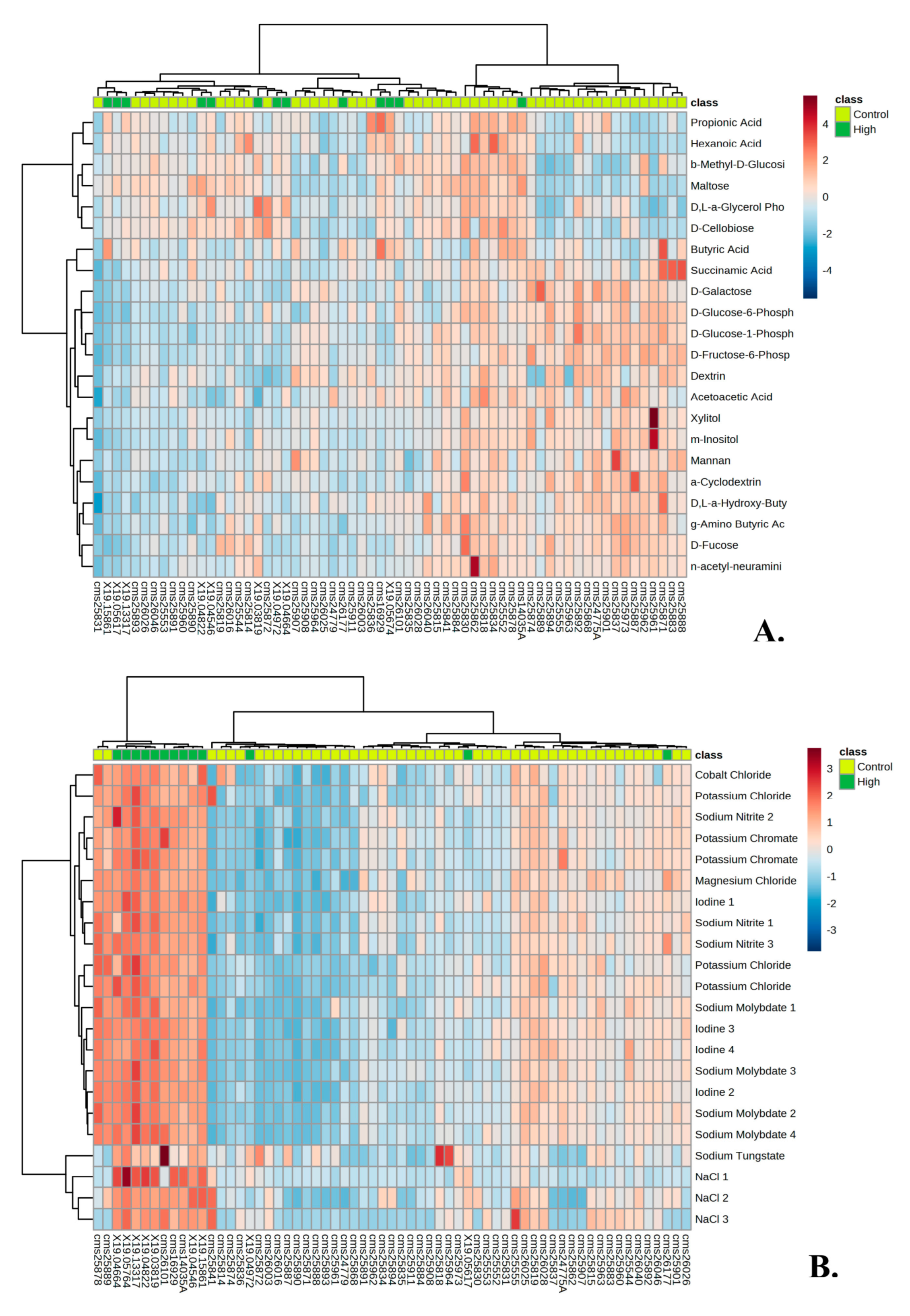
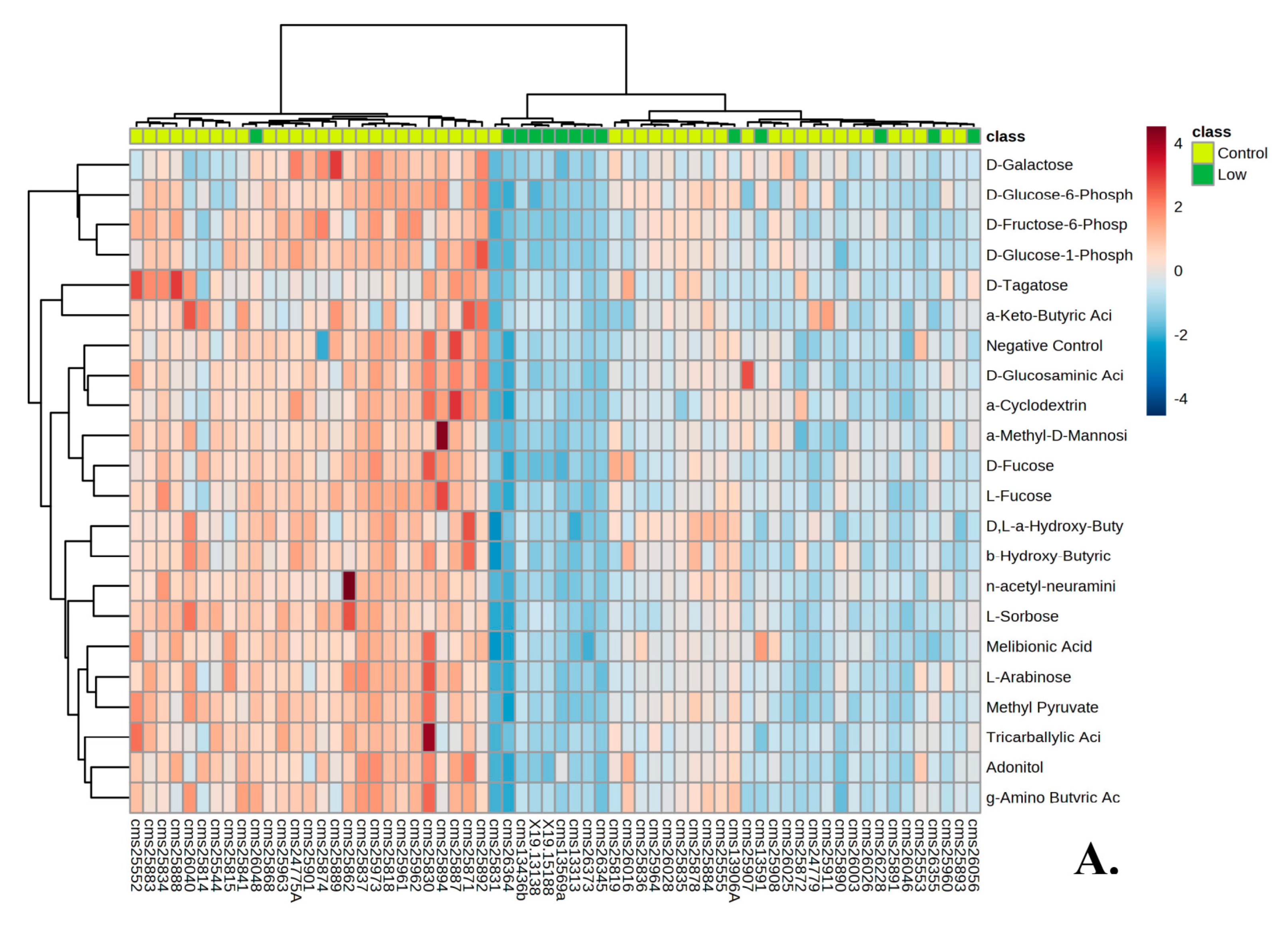
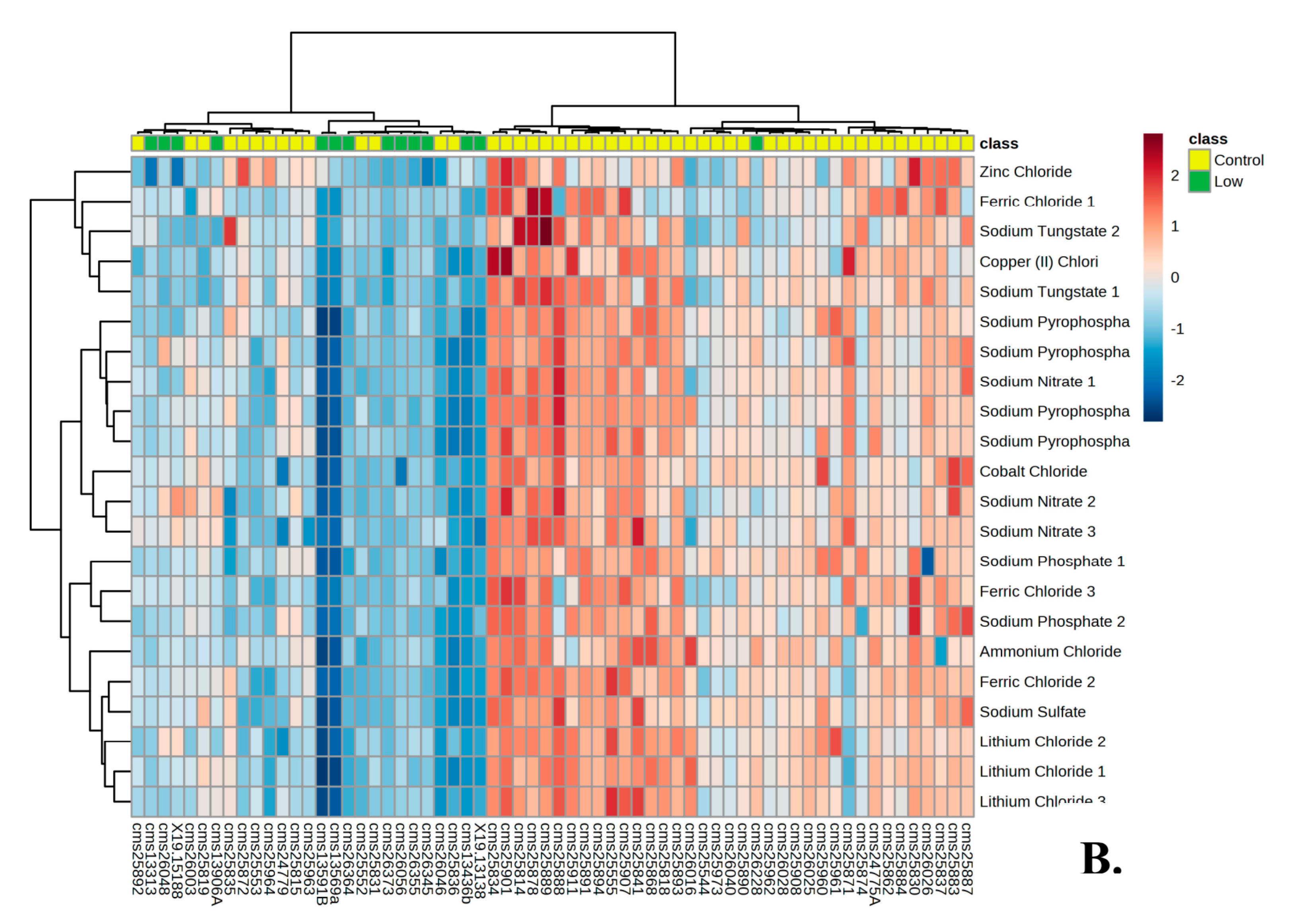
3.1. Metabolic Profile of Individuals with PMS
3.2. Metabolic Profile of High and Low Responders
3.3. Response to hGH and IGF-1 in Comparison to Baseline Glucose Metabolism
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Phelan, K.; Rogers, R.C.; Boccuto, L. Phelan-McDermid Syndrome; Adam, M., Ardinger, H., Pagon, R., Eds.; University of Washington: Seattle, WA, USA, 2005. [Google Scholar]
- Costales, J.L.; Kolevzon, A. Phelan–McDermid Syndrome and SHANK3: Implications for Treatment. Neurotherapeutics 2015, 12, 620–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolevzon, A.; Bush, L.; Wang, A.T.; Halpern, D.; Frank, Y.; Grodberg, D.; Rapaport, R.; Tavassoli, T.; Chaplin, W.; Soorya, L.; et al. A pilot controlled trial of insulin-like growth factor-1 in children with Phelan-McDermid syndrome. Mol. Autism 2014, 5, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabrucker, A.M.; Schmeisser, M.J.; Schoen, M.; Boeckers, T.M. Postsynaptic ProSAP/Shank scaffolds in the cross-hair of synaptopathies. Trends Cell Biol. 2011, 21, 594–603. [Google Scholar] [CrossRef] [PubMed]
- Oberman, L.M.; Boccuto, L.; Cascio, L.; Sarasua, S.; Kaufmann, W.E. Autism spectrum disorder in Phelan-McDermid syn-drome: Initial characterization and genotype-phenotype correlations. Orphanet. J. Rare Dis. 2015, 10, 105. [Google Scholar] [CrossRef] [Green Version]
- Bozdagi, O.; Sakurai, T.; Papapetrou, D.; Wang, X.; Dickstein, D.L.; Takahashi, N.; Kajiwara, Y.; Yang, M.; Katz, A.M.; Scattoni, M.L.; et al. Haploinsufficiency of the autism-associated Shank3 gene leads to deficits in synaptic function, social interaction, and social communication. Mol. Autism 2010, 1, 15. [Google Scholar] [CrossRef] [Green Version]
- Kolevzon, A.; Angarita, B.; Bush, L.; Wang, A.T.; Frank, Y.; Yang, A.; Rapaport, R.; Saland, J.; Srivastava, S.; Farrell, C.; et al. Phelan-McDermid syndrome: A review of the literature and practice parameters for medical assessment and monitoring. J. Neurodev. Disord. 2014, 6, 39. [Google Scholar] [CrossRef]
- Phelan, K.; McDermid, H. The 22q13.3 Deletion Syndrome (Phelan-McDermid Syndrome). Mol. Syndr. 2011, 2, 186–201. [Google Scholar] [CrossRef] [Green Version]
- Sarasua, S.M.; Dwivedi, A.; Boccuto, L.; Rollins, J.D.; Chen, C.-F.; Rogers, R.C.; Phelan, K.; DuPont, B.R.; Collins, J.S. Association between deletion size and important phenotypes expands the genomic region of interest in Phelan-McDermid syndrome (22q13 deletion syndrome). J. Med. Genet. 2011, 48, 761–766. [Google Scholar] [CrossRef]
- Alexandrov, P.N.; Zhao, Y.; Jaber, V.; Cong, L.; Lukiw, W.J. Deficits in the Proline-Rich Synapse-Associated Shank3 Protein in Multiple Neuropsychiatric Disorders. Front. Neurol. 2017, 8, 670. [Google Scholar] [CrossRef] [Green Version]
- Mei, Y.; Monteiro, P.; Zhou, Y.; Kim, J.-A.; Gao, X.; Fu, Z.; Feng, G. Adult restoration of Shank3 expression rescues selective autistic-like phenotypes. Nature 2016, 530, 481–484. [Google Scholar] [CrossRef]
- Pchitskaya, E.; Bezprozvanny, I. Dendritic Spines Shape Analysis—Classification or Clusterization? Perspective. Front. Synaptic Neurosci. 2020, 12, 31. [Google Scholar] [CrossRef]
- Bozdagi, O.; Tavassoli, T.; Buxbaum, J.D. Insulin-like growth factor-1 rescues synaptic and motor deficits in a mouse model of autism and developmental delay. Mol. Autism 2013, 4, 9. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.-H.; Ehlers, M.D. Modeling Autism by SHANK Gene Mutations in Mice. Neuron 2013, 78, 8–27. [Google Scholar] [CrossRef] [Green Version]
- Monteiro, P.; Feng, G. SHANK proteins: Roles at the synapse and in autism spectrum disorder. Nat. Rev. Neurosci. 2017, 18, 147–157. [Google Scholar] [CrossRef]
- Peça, J.; Feliciano, C.; Ting, J.T.; Wang, W.; Wells, M.F.; Venkatraman, T.N.; Lascola, C.D.; Fu, Z.; Feng, G. Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature 2011, 472, 437–442. [Google Scholar] [CrossRef] [Green Version]
- Fischer, I.; Shohat, S.; Levy, G.; Bar, E.; Trangle, S.S.; Efrati, S.; Barak, B. Hyperbaric Oxygen Therapy Alleviates Social Behavior Dysfunction and Neuroinflammation in a Mouse Model for Autism Spectrum Disorders. Int. J. Mol. Sci. 2022, 23, 11077. [Google Scholar] [CrossRef] [PubMed]
- Sethuram, S.; Levy, T.; Foss-Feig, J.; Halpern, D.; Sandin, S.; Siper, P.M.; Walker, H.; Buxbaum, J.D.; Rapaport, R.; Kolevzon, A. A proof-of-concept study of growth hormone in children with Phelan–McDermid syndrome. Mol. Autism 2022, 13, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Kolevzon, A.; Breen, M.S.; Siper, P.M.; Halpern, D.; Frank, Y.; Rieger, H.; Weismann, J.; Trelles, M.P.; Lerman, B.; Rapaport, R.; et al. Clinical trial of insulin-like growth factor-1 in Phelan-McDermid syndrome. Mol. Autism 2022, 13, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Vahdatpour, C.; Dyer, A.; Tropea, D. Insulin-Like Growth Factor 1 and Related Compounds in the Treatment of Childhood-Onset Neurodevelopmental Disorders. Front. Neurosci. 2016, 10, 450. [Google Scholar] [CrossRef] [Green Version]
- Shcheglovitov, A.; Shcheglovitova, O.; Yazawa, M.; Portmann, T.; Shu, R.; Sebastiano, V.; Krawisz, A.; Froehlich, W.; Bernstein, J.A.; Hallmayer, J.F.; et al. SHANK3 and IGF1 restore synaptic deficits in neurons from 22q13 deletion syndrome patients. Nature 2013, 503, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Xie, R.J.; Li, T.X.; Sun, C.; Cheng, C.; Zhao, J.; Xu, H.; Liu, Y. A case report of Phelan-McDermid syndrome: Preliminary results of the treatment with growth hormone therapy. Ital. J. Pediatr. 2021, 47, 1–4. [Google Scholar] [CrossRef]
- Zwanenburg, R.J.; Bocca, G.; Ruiter, S.A.; Dillingh, J.H.; Flapper, B.C.; van den Heuvel, E.R.; van Ravenswaaij-Arts, C. Is there an effect of intranasal insulin on development and behaviour in Phelan-McDermid syndrome?: A randomized, double-blind, placebo-controlled trial. Eur. J. Hum. Genet. 2016, 24, 1696–1701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, L.; Oberman, L.M.; Beamer, L.; Cascio, L.; May, M.; Srikanth, S.; Skinner, C.; Jones, K.; Allen, B.; Rogers, C.; et al. Genetic and metabolic profiling of individuals with Phelan-McDermid syn-drome presenting with seizures. Clin. Genet. 2022, 101, 87–100. [Google Scholar] [CrossRef] [PubMed]
- Srikanth, S.; Jain, L.; Zepeda-Mendoza, C.; Cascio, L.; Jones, K.; Pauly, R.; DuPont, B.; Rogers, C.; Sarasua, S.; Phelan, K.; et al. Position effects of 22q13 rearrangements on candidate genes in Phelan-McDermid syndrome. PLoS ONE 2021, 16, e0253859. [Google Scholar] [CrossRef] [PubMed]
- Boccuto, L.; Chen, C.-F.; Pittman, A.R.; Skinner, C.D.; McCartney, H.J.; Jones, K.; Bochner, B.R.; Stevenson, R.E.; Schwartz, C.E. Decreased tryptophan metabolism in patients with autism spectrum disorders. Mol. Autism 2013, 4, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cascio, L.; Chen, C.; Pauly, R.; Srikanth, S.; Jones, K.; Skinner, C.D.; Stevenson, R.E.; Schwartz, C.E.; Boccuto, L. Abnormalities in the genes that encode Large Amino Acid Transporters increase the risk of Autism Spectrum Disorder. Mol. Genet. Genom. Med. 2019, 8, e1036. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Differential Compounds | Difference in NADH between Individuals with PMS and Controls (p-Values) | Differential Compounds | Difference in NADH between Individuals with PMS and Controls (p-Values) |
|---|---|---|---|
| PM–M1: | |||
| B2: D-Glucose-1-Phosphate |  1 × 10−5 1 × 10−5 | A3: Negative Control | 0.00876 |
| D6: D-Fructose-6-Phosphate |  1 × 10−5 1 × 10−5 | A1: Negative Control | 0.0094 |
| B1: D-Glucose-6-Phosphate | 0.00007 | A9: Maltose |  0.0094 0.0094 |
| E3: D-Galactose | 0.00013 | F12: Citric Acid |  0.01144 0.01144 |
| D5: D-Fucose | 0.00015 | E4: a-Methyl-D-Galactoside | 0.0116 |
| C1: D-Glucosaminic Acid | 0.00061 | H1: Acetoacetic Acid | 0.01346 |
| H4: D,L-a-Hydroxy-Butyric Acid | 0.00102 | C3: Chondroitin Sulfate | 0.01663 |
| H5: b-Hydroxy-Butyric Acid | 0.00232 | E5: b-Methyl-D-Galactoside | 0.01724 |
| H10: Propionic Acid | 0.00265 | A2: Negative Control | 0.01906 |
| A5: Dextrin | 0.0029 | A11: D-Cellobiose | 0.0205 |
| E6: n-acetyl-neuraminic acid | 0.00464 | B3: L-Glucose | 0.0205 |
| B4: D-(+)-Glucose | 0.0061 | D3: L-Rhamnose | 0.02218 |
| B5: D-(+)-Glucose | 0.0061 | C12: Turanose | 0.02388 |
| C6: a-Methyl-D-Mannoside | 0.0061 | D2: L-Sorbose | 0.02388 |
| E1: Melibionic Acid | 0.0061 | D1: D-Tagatose | 0.02581 |
| F1: Adonitol | 0.0061 | E11: Adenosine | 0.02603 |
| F6: m-Inositol | 0.0061 | E12: Inosine | 0.02603 |
| G5: Pyruvic Acid | 0.0061 | F2: L-Arabinose | 0.02603 |
| H2: g-Amino Butyric Acid | 0.0061 | F4: b-Methyl-D-Xyloside | 0.02603 |
| A4: a-Cyclodextrin | 0.00676 | G8: Succinic Acid | 0.02603 |
| C4: Mannan | 0.00676 | H7: Butyric Acid | 0.0276 |
| C5: D-Mannose | 0.00676 | B10: Salicin | 0.03039 |
| D4: L-Fucose | 0.00676 | B11: D-Sorbitol | 0.03039 |
| F5: Xylitol | 0.00676 | G1: Tricarballylic Acid | 0.03039 |
| G7: Succinamic Acid | 0.00676 | E2: D-Melibiose | 0.03823 |
| H3: a-Keto-Butyric Acid | 0.00676 | G4: Methyl Pyruvate | 0.03823 |
| H12: Hexanoic Acid | 0.00717 | A6: Glycogen | 0.04949 |
| PM–M2: | |||
| E1: Ala-Pro | 0.00446 | C8: D-Threonine | 0.02213 |
| F1: Arg-Leu | 0.00446 | D6: Ala-Gly | 0.02213 |
| H4: Glu-Val | 0.00446 | F2: Arg-Lys | 0.02213 |
| D1: Ala-Arg | 0.00491 | F11: Asp-Ala | 0.02213 |
| B1: L-Aspartic Acid | 0.00708 | G9: Glu-Ala | 0.02213 |
| E7: Arg-Ala | 0.00708 | H1: Glu-Ser | 0.02213 |
| F4: Arg-Phe | 0.00708 | F7: Arg-Tyr | 0.02334 |
| D9: Ala-Leu | 0.00735 | G5: Asp-Lys | 0.02847 |
| G1: Asp-Glu | 0.00735 | D4: Ala-Glu | 0.03188 |
| H2: Glu-Trp | 0.00862 | E6: Ala-Val | 0.03495 |
| D7: Ala-His | 0.01228 | C6: D-Serine | 0.04029 |
| E4: Ala-Trp | 0.0135 | H8: Gly-Ala | 0.04029 |
| E3: Ala-Thr | 0.01606 | D12: Ala-Phe | 0.04078 |
| G8: Asp-Val | 0.01606 | A1: Negative Control | 0.04624 |
| D5: Ala-Gln | 0.01718 | A8: L-Alaninamide | 0.04624 |
| F5: Arg-Ser | 0.01718 | B3: L-Glutamic Acid | 0.04624 |
| G4: Asp-Leu | 0.01744 | D10: Ala-Lys | 0.04624 |
| F6: Arg-Trp | 0.01814 | G11: Glu-Glu | 0.04624 |
| A3: Negative Control | 0.02156 | F3: Arg-Met | 0.04674 |
| D8: Ala-Ile | 0.02156 | G2: Asp-Gln | 0.04674 |
| G6: Asp-Phe | 0.02156 | A2: Negative Control | 0.04747 |
| G7: Asp-Trp | 0.02207 | ||
| PM–M3: | |||
| B1: Gly-Thr | 0.01047 | E7: Leu-Phe | 0.04043 |
| C4: His-Tyr | 0.01228 | F1: Lys-Ala | 0.04043 |
| D1: Ile-Leu | 0.0157 | F10: Lys-Phe | 0.04043 |
| A8: Gly-Lys | 0.02377 | C3: His-Trp | 0.0411 |
| C6: Ile-Ala | 0.02377 | D9: Leu-Ala | 0.0411 |
| C1: His-Pro | 0.04043 | E8: Leu-Pro | 0.0411 |
| D4: Ile-Pro | 0.04043 | A10: Gly-Phe | 0.04198 |
| D6: Ile-Trp | 0.04043 | E6: Leu-Met | 0.04583 |
| E5: Leu-Leu | 0.04043 | D5: Ile-Ser | 0.04805 |
| PM–M5: | |||
| A9: NaCl | 1 × 10−5 | G9: Sodium Nitrite | 0.00196 |
| A8: NaCl | 0.00001 | E11: Sodium Molybdate | 0.00286 |
| A7: NaCl | 0.00002 | F12: Potassium Chromate | 0.00366 |
| B11: Potassium Chloride | 0.00018 | H10: Magnesium Chloride | 0.00366 |
| F4: Sodium Tungstate | 0.00018 | F9: Potassium Chromate | 0.00623 |
| F2: Sodium Tungstate | 0.00052 | G11: Sodium Nitrite | 0.00644 |
| D10: Iodine | 0.00074 | H11: Magnesium Chloride | 0.00974 |
| B10: Potassium Chloride | 0.00077 | F8: Sodium Orthovanadate | 0.0103 |
| D9: Iodine | 0.00089 | G1: Sodium Pyrophosphate | 0.01031 |
| B9: Potassium Chloride | 0.00111 | H9: Magnesium Chloride | 0.01031 |
| E10: Sodium Molybdate | 0.00111 | C8: Manganese Chloride | 0.01134 |
| E12: Sodium Molybdate | 0.00111 | D8: Cobalt Chloride | 0.01134 |
| F1: Sodium Tungstate | 0.00111 | D1: Copper (II) Chloride | 0.01536 |
| F11: Potassium Chromate | 0.00123 | G3: Sodium Pyrophosphate | 0.017 |
| G10: Sodium Nitrite | 0.00123 | H12: Magnesium Chloride | 0.02089 |
| G12: Sodium Nitrite | 0.00123 | D5: Cobalt Chloride | 0.03199 |
| F10: Potassium Chromate | 0.0014 | D4: Copper (II) Chloride | 0.0427 |
| D12: Iodine | 0.00161 | G4: Sodium Pyrophosphate | 0.0458 |
| E9: Sodium Molybdate | 0.00161 | C12: Zinc Chloride | 0.04771 |
| D11: Iodine | 0.00171 | ||
| PM–M6: | |||
| A12: Dibutyryl-cAMP | 0.01099 | D2: L-Leucine | 0.02157 |
| B1: 3-Isobutyl-1-Methylxanthine | 0.01099 | D4: L-Leucine | 0.02157 |
| B2: 3-Isobutyl-1-Methylxanthine | 0.01099 | F2: Dexamethasone | 0.02253 |
| B4: 3-Isobutyl-1-Methylxanthine | 0.01099 | C12: Norepinephrine | 0.02864 |
| C1: Epinephrine | 0.01099 | E4: Triiodothyronine | 0.02864 |
| D1: L-Leucine | 0.01099 | F3: Dexamethasone | 0.0306 |
| E1: Triiodothyronine | 0.01099 | D12: Creatine | 0.03105 |
| F1: Dexamethasone | 0.01099 | C2: Epinephrine | 0.03287 |
| A11: Dibutyryl-cAMP | 0.01103 | C4: Epinephrine | 0.03376 |
| B3: 3-Isobutyl-1-Methylxanthine | 0.01103 | A1: Negative Control | 0.03388 |
| B10: Caffeine | 0.01537 | A9: Dibutyryl-cAMP | 0.03388 |
| E3: Triiodothyronine | 0.01537 | C11: Norepinephrine | 0.03388 |
| A10: Dibutyryl-cAMP | 0.02017 | A2: Negative Control | 0.03607 |
| B11: Caffeine | 0.02017 | B9: Caffeine | 0.03607 |
| D3: L-Leucine | 0.02017 | C5: Epinephrine | 0.03607 |
| F4: Dexamethasone | 0.02017 | F12: Hydrocortisone | 0.03607 |
| G1: Progesterone | 0.02017 | A2: Negative Control | 0.03652 |
| B6: 3-Isobutyl-1-Methylxanthine | 0.02157 | G5: Progesterone | 0.03748 |
| C3: Epinephrine | 0.02157 | H9: Aldosterone | 0.03748 |
| C6: Epinephrine | 0.02157 | F6: Dexamethasone | 0.03951 |
| PM–M7: | |||
| A12: Insulin | 0.01964 | A10: Insulin | 0.02473 |
| B11: Glucagon | 0.02006 | B12: Glucagon | 0.02765 |
| A11: Insulin | 0.02088 | C10: Leptin | 0.03238 |
| B10: Glucagon | 0.02088 | C11: Leptin | 0.03238 |
| B9: Glucagon | 0.02437 | C12: Leptin | 0.03238 |
| TRP: | |||
| A1: a-D-Glucose | 1 × 10−5 | E1: Trp-Lys | 0.00023 |
| D1: Trp-Gly | 0.00003 | F1: Trp-Ala | 0.00067 |
| C1: L-Tryptophan | 0.00005 | H1: Trp-Leu | 0.00357 |
| hGH High Responders | % | IGF-1 High Responders | % | # Wells in Common | |
| PM–M1: | 3 | 3.13% | 3 | 3.13% | 3 |
| PM–M2: | 0 | 0.00% | 0 | 0.00% | 0 |
| PM–M3: | 0 | 0.00% | 0 | 0.00% | 0 |
| PM–M4: | 0 | 0.00% | 0 | 0.00% | 0 |
| PM–M5: | 39 | 40.63% | 43 | 44.79% | 39 |
| PM–M6: | 63 | 65.63% | 62 | 64.58% | 59 |
| PM–M7: | 93 | 96.88% | 91 | 94.79% | 91 |
| PM–M8: | 89 | 92.71% | 89 | 92.71% | 89 |
| TRP: | 3 | 37.50% | 5 | 62.50% | 3 |
| hGH Low responders | % | IGF-1 Low responders | % | # Wells in common | |
| PM–M1: | 52 | 54.17% | 49 | 51.04% | 48 |
| PM–M2: | 60 | 62.50% | 66 | 68.75% | 55 |
| PM–M3: | 71 | 73.96% | 62 | 64.58% | 60 |
| PM–M4: | 0 | 0.00% | 0 | 0.00% | 0 |
| PM–M5: | 50 | 52.08% | 49 | 51.04% | 48 |
| PM–M6: | 85 | 88.54% | 88 | 91.67% | 85 |
| PM–M7: | 95 | 98.96% | 95 | 98.96% | 95 |
| PM–M8: | 96 | 100.00% | 96 | 100.00% | 96 |
| TRP: | 5 | 62.50% | 2 | 25.00% | 2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moffitt, B.A.; Sarasua, S.M.; Ivankovic, D.; Ward, L.D.; Valentine, K.; Bennett, W.E., Jr.; Rogers, C.; Phelan, K.; Boccuto, L. Stratification of a Phelan–McDermid Syndrome Population Based on Their Response to Human Growth Hormone and Insulin-like Growth Factor. Genes 2023, 14, 490. https://doi.org/10.3390/genes14020490
Moffitt BA, Sarasua SM, Ivankovic D, Ward LD, Valentine K, Bennett WE Jr., Rogers C, Phelan K, Boccuto L. Stratification of a Phelan–McDermid Syndrome Population Based on Their Response to Human Growth Hormone and Insulin-like Growth Factor. Genes. 2023; 14(2):490. https://doi.org/10.3390/genes14020490
Chicago/Turabian StyleMoffitt, Bridgette A., Sara M. Sarasua, Diana Ivankovic, Linda D. Ward, Kathleen Valentine, William E. Bennett, Jr., Curtis Rogers, Katy Phelan, and Luigi Boccuto. 2023. "Stratification of a Phelan–McDermid Syndrome Population Based on Their Response to Human Growth Hormone and Insulin-like Growth Factor" Genes 14, no. 2: 490. https://doi.org/10.3390/genes14020490