
Pantothenate and L-Carnitine Supplementation Improves Pathological Alterations in Cellular Models of KAT6A Syndrome
 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Ethical Statements
2.3. Cell Culture
2.4. Immunoblotting
2.5. Drug Screening
2.6. Immunofluorescence Microscopy
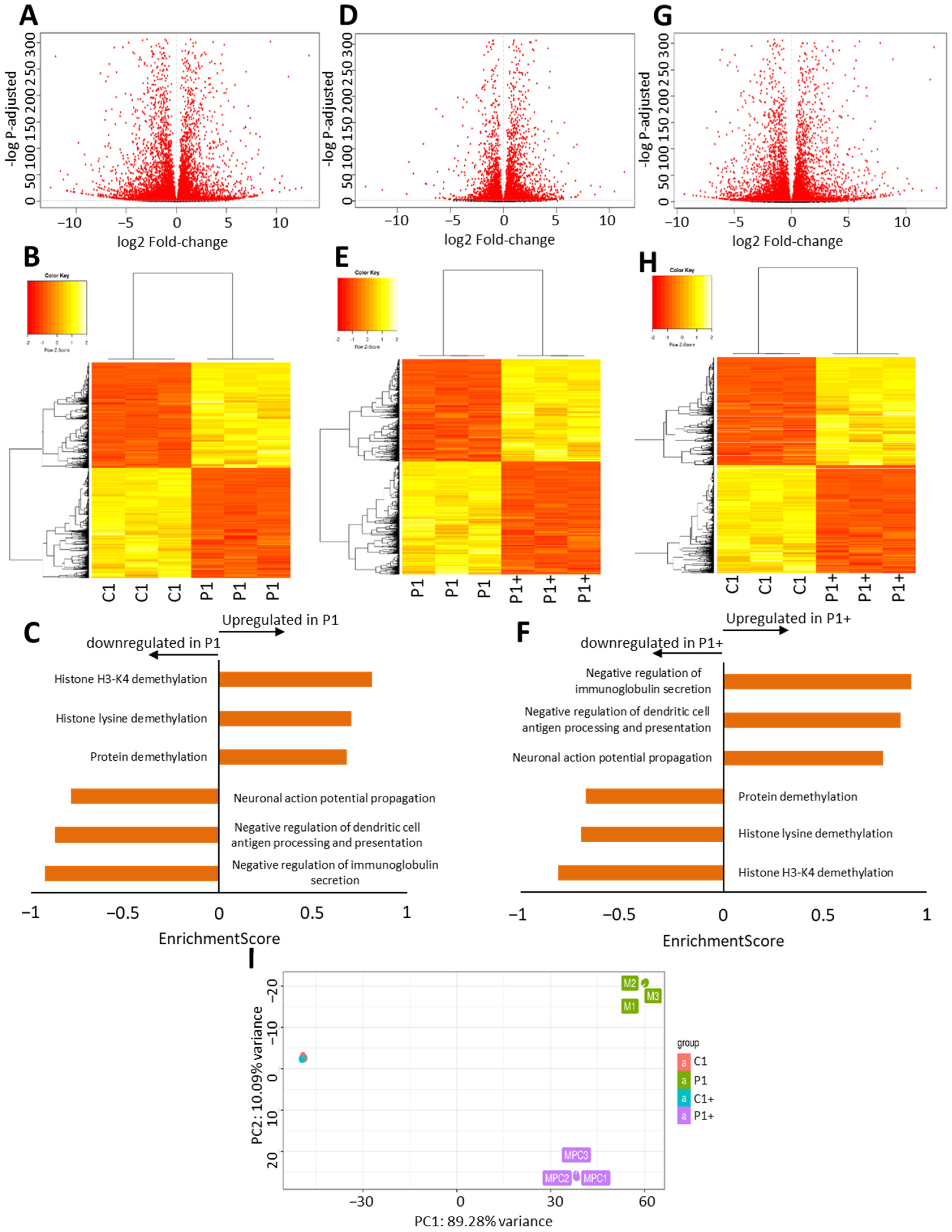
2.7. RNAseq
2.8. Cell Fractioning
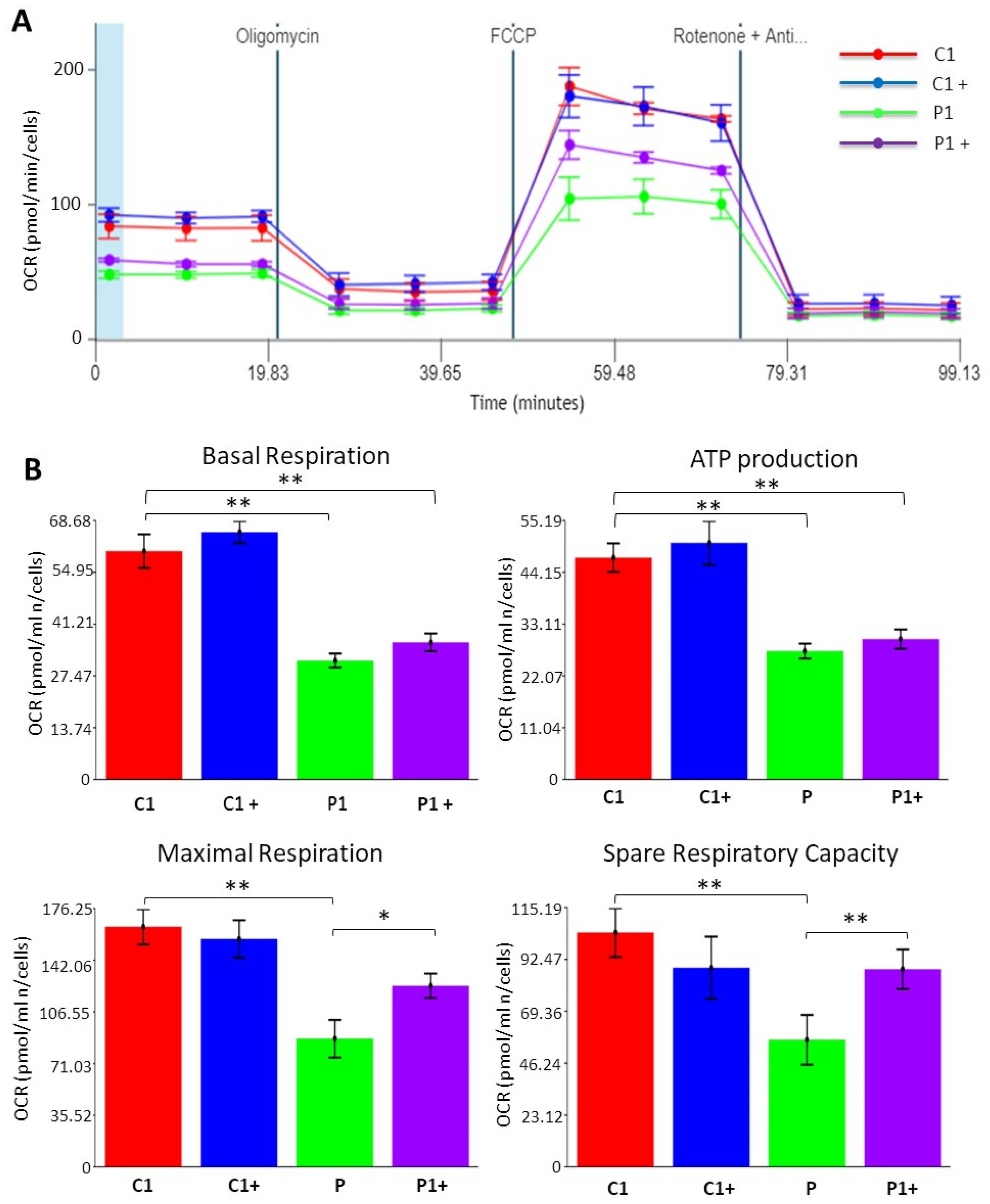
2.9. Bioenergetics
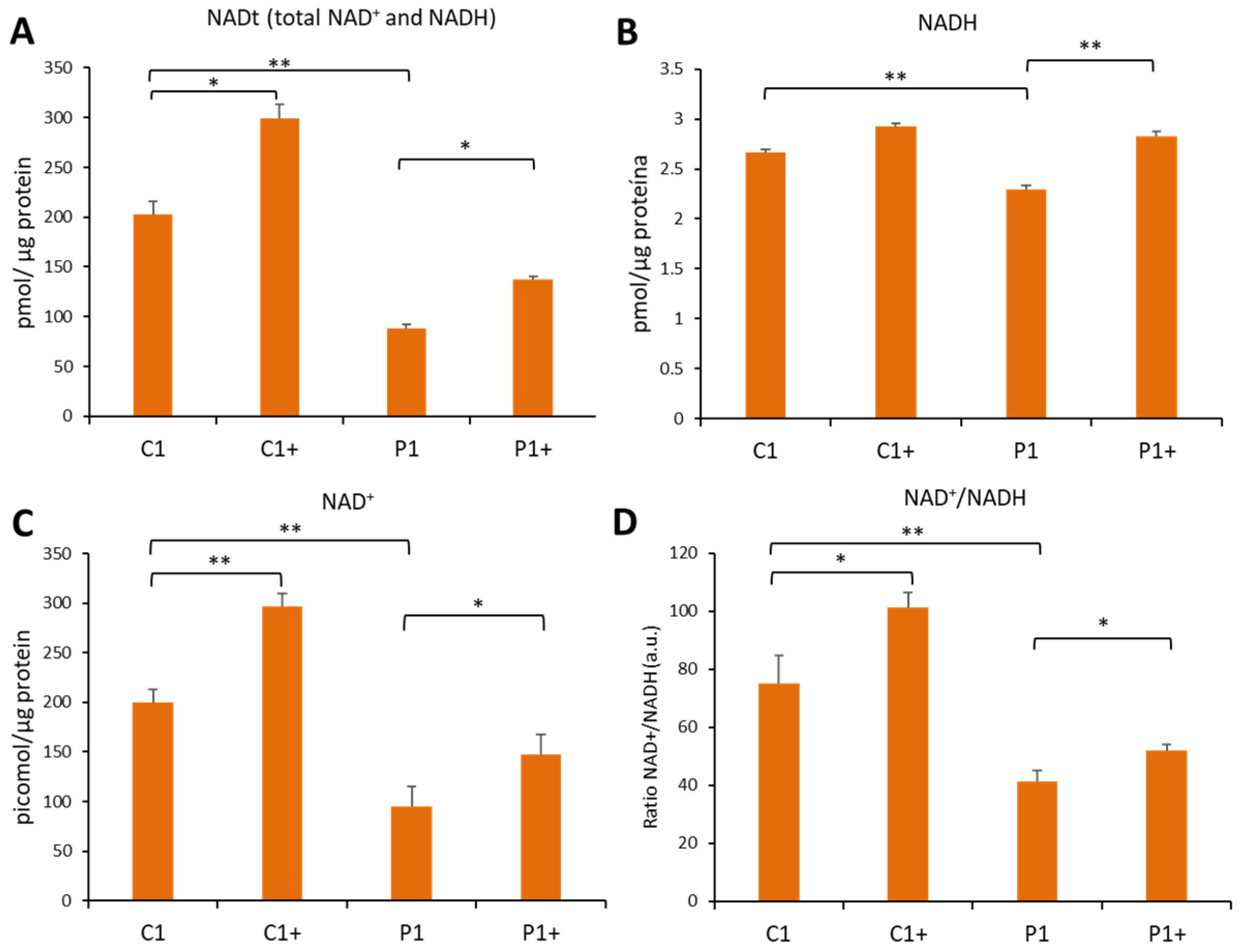
2.10. NAD+/NADH Levels
2.11. Histone H3 Total Acetylation
2.12. Statistical Analyses
3. Results
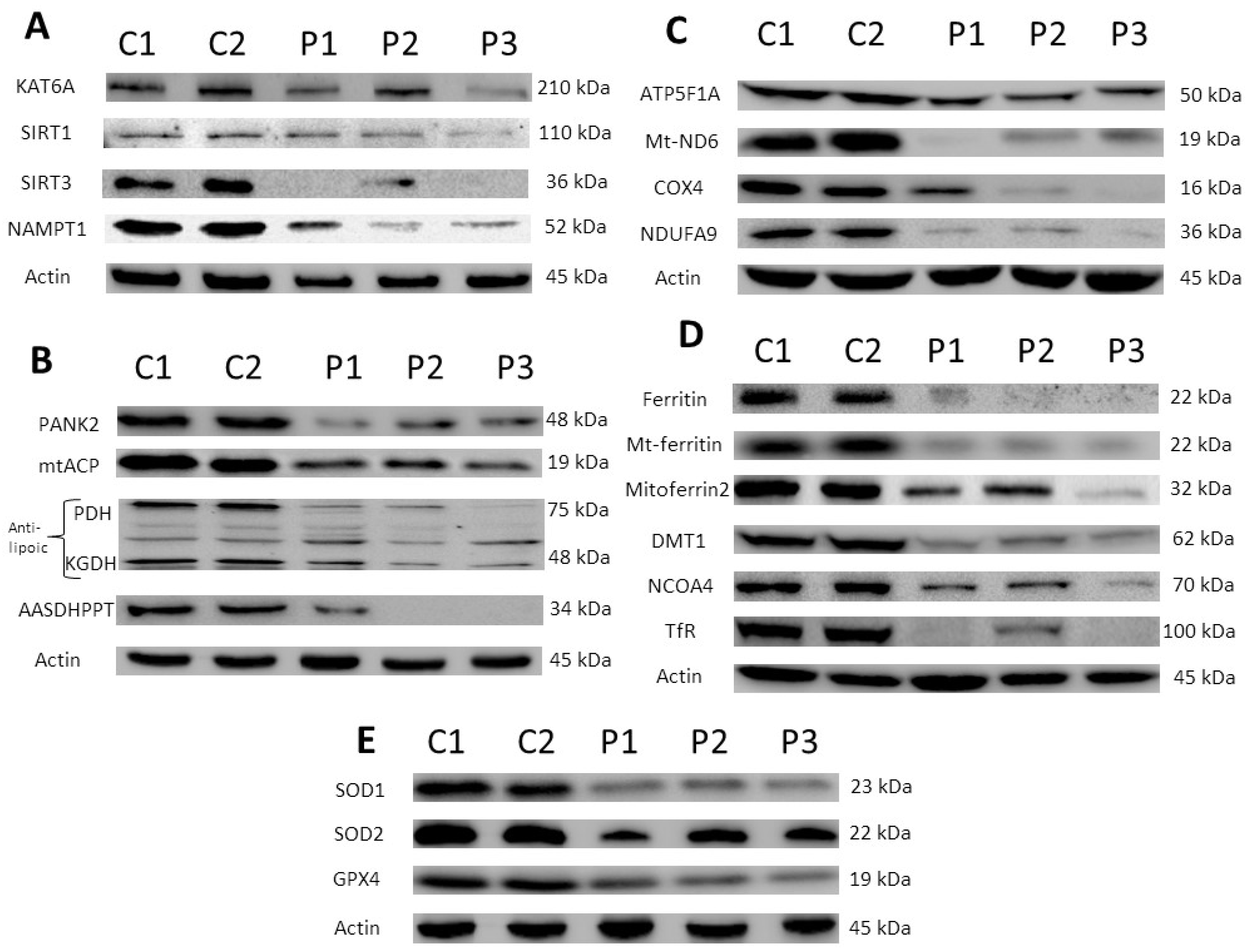
3.1. Protein Expression Levels in KAT6A Fibroblasts
3.1.1. Expression Levels of the KAT6A Enzyme Are Markedly Reduced in Fibroblasts Derived from KAT6A Patients
3.1.2. Expression of Proteins Involved in Coenzyme A (CoA) Metabolism Were Also Affected in Mutant KAT6A Fibroblasts
3.1.3. Expression Levels of Mitochondrial Respiratory Chain Proteins Were Affected in KAT6A Mutant Fibroblasts
3.1.4. Expression Levels of Several Proteins Implicated in Iron Metabolism Were Reduced in Mutant KAT6A Fibroblasts
3.1.5. Expression Levels of Antioxidant Enzymes Were Also Reduced in Mutant KAT6A Fibroblasts
3.2. Effect of Pantothenate on KAT6A Fibroblasts
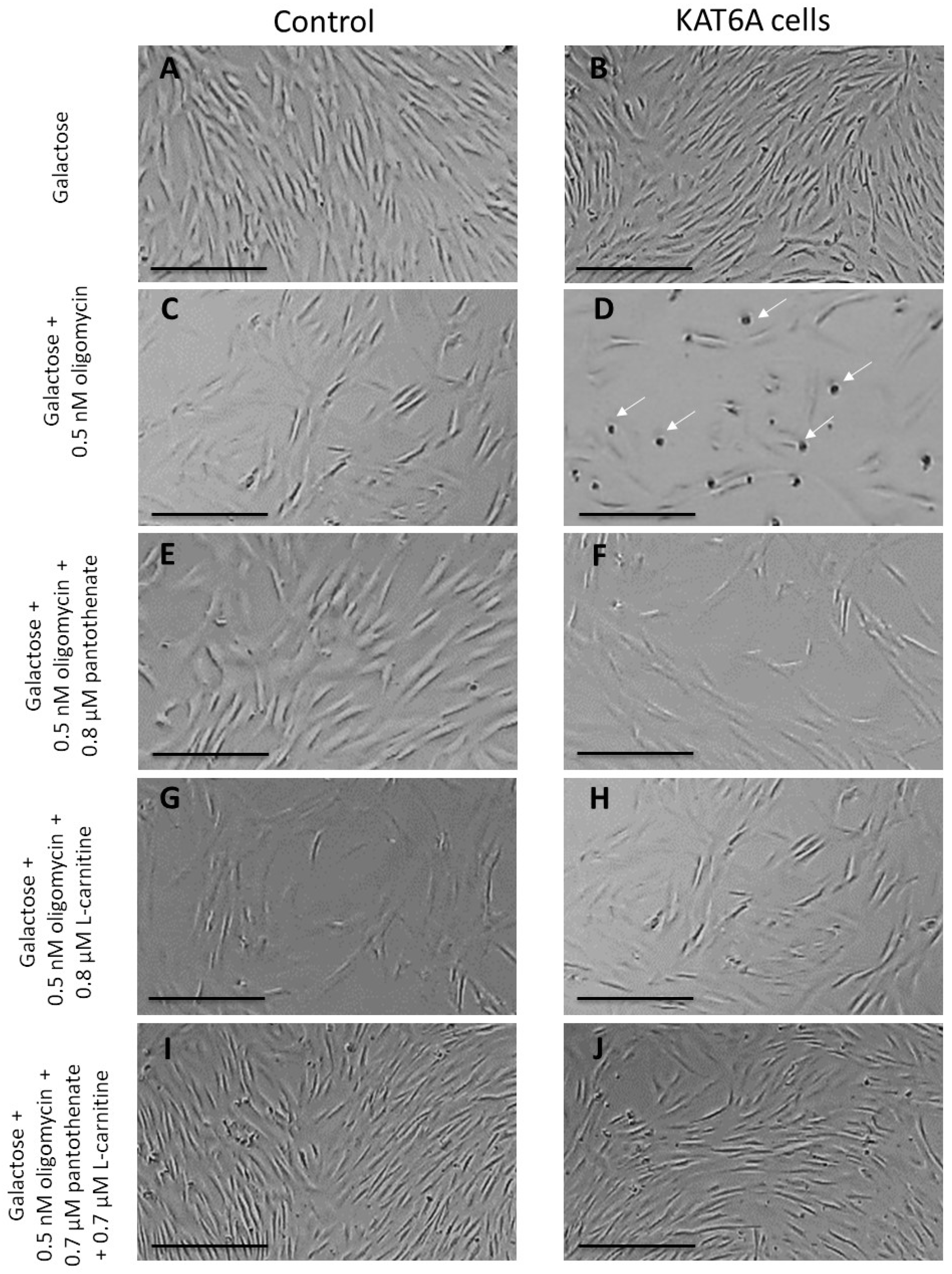
3.2.1. Pantothenate and L-Carnitine Supplementation Enhance the Survival of the Mutant KAT6A Fibroblasts in Nutritional Stress Medium
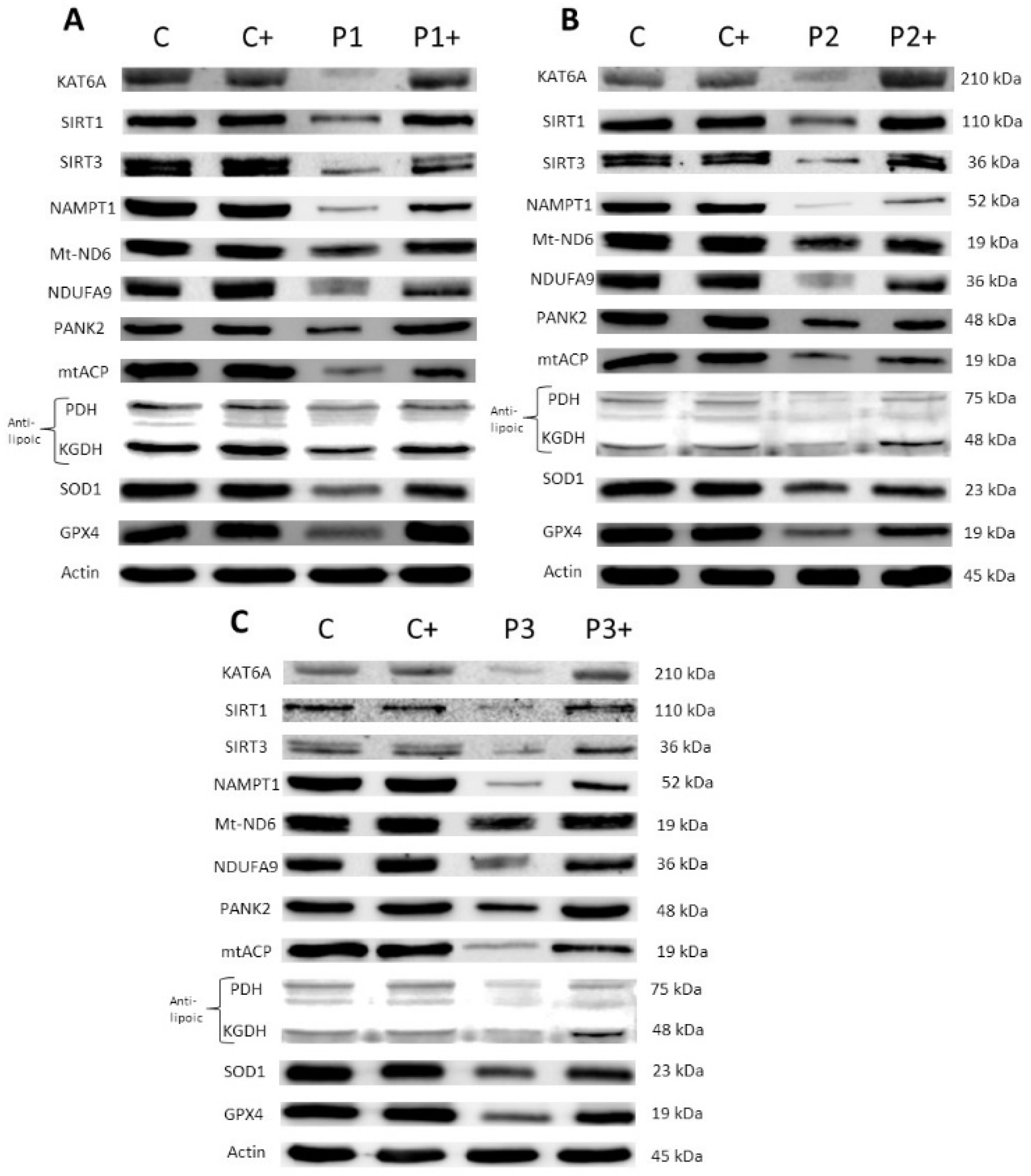
3.2.2. Pantothenate and L-Carnitine Supplementation Partially Correct Protein Expression Patterns in Mutant KAT6A Cell Lines
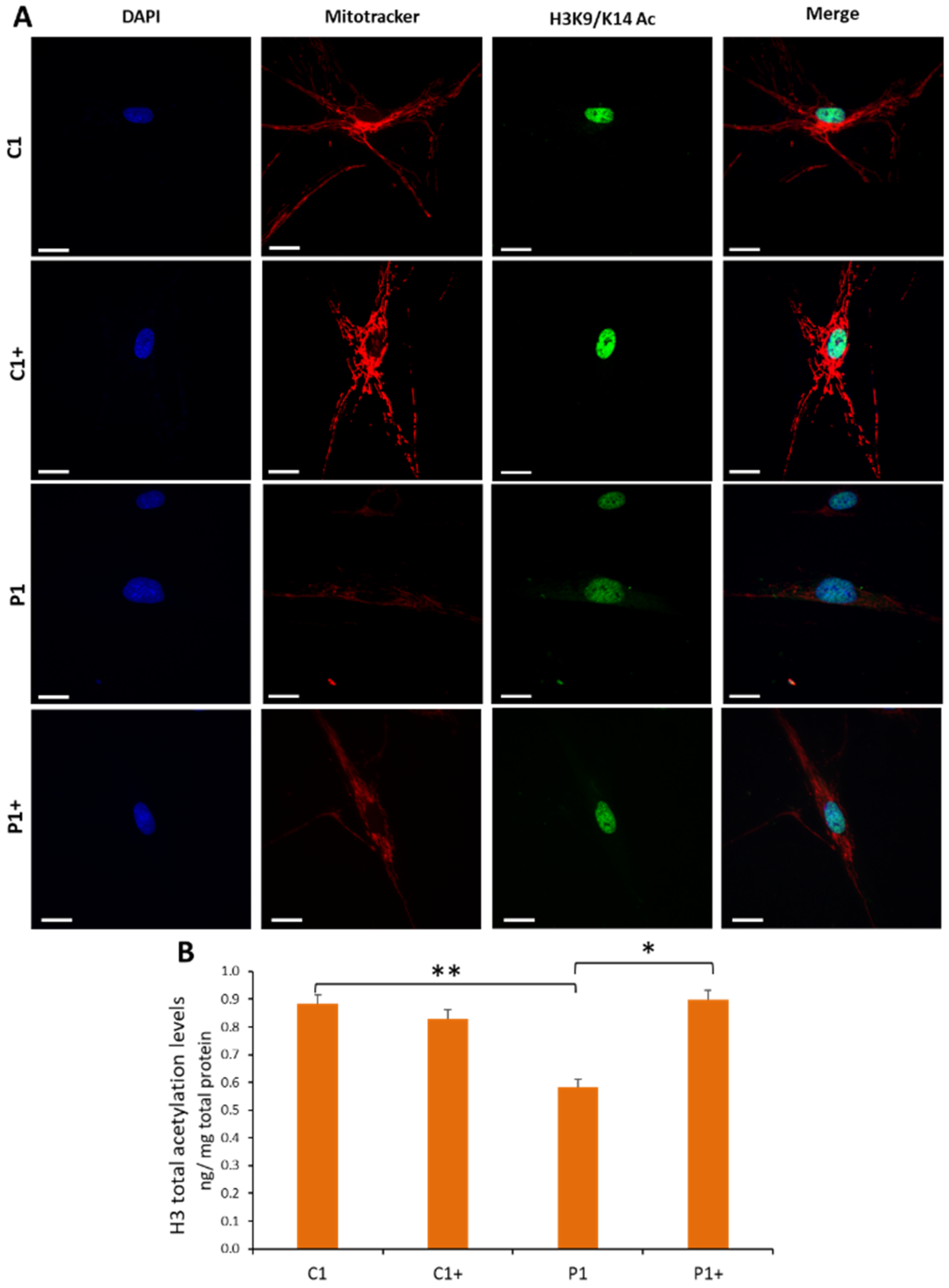
3.2.3. Pantothenate and L-Carnitine Supplementation Increases Histones Acetylation in KAT6A Cells
3.2.4. Pantothenate and L-Carnitine Supplementation Improves Cell Bioenergetics in KAT6A Mutant Fibroblasts
3.2.5. Pantothenate and L-Carnitine Treatment Highly Modifies the Transcriptome
4. Discussion
Pantothenate and L-Carnitine as Epigenetic Modulators
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Kennedy, J.; Goudie, D.; Blair, E.; Chandler, K.; Joss, S.; McKay, V.; Green, A.; Armstrong, R.; Lees, M.; Kamien, B.; et al. KAT6A Syndrome: Genotype-phenotype correlation in 76 patients with pathogenic KAT6A variants. Genet. Med. 2019, 21, 850–860. [Google Scholar] [CrossRef]
- Lee, H.; Deignan, J.L.; Dorrani, N.; Strom, S.P.; Kantarci, S.; Quintero-Rivera, F.; Das, K.; Toy, T.; Harry, B.; Yourshaw, M.; et al. Clinical exome sequencing for genetic identification of rare Mendelian disorders. JAMA 2014, 312, 1880–1887. [Google Scholar] [CrossRef] [PubMed]
- Arboleda, V.A.; Lee, H.; Dorrani, N.; Zadeh, N.; Willis, M.; Macmurdo, C.F.; Manning, M.A.; Kwan, A.; Hudgins, L.; Barthelemy, F.; et al. De novo nonsense mutations in KAT6A, a lysine acetyl-transferase gene, cause a syndrome including microcephaly and global developmental delay. Am. J. Hum. Genet. 2015, 96, 498–506. [Google Scholar] [CrossRef] [Green Version]
- Tham, E.; Lindstrand, A.; Santani, A.; Malmgren, H.; Nesbitt, A.; Dubbs, H.A.; Zackai, E.H.; Parker, M.J.; Millan, F.; Rosenbaum, K.; et al. Dominant mutations in KAT6A cause intellectual disability with recognizable syndromic features. Am. J. Hum. Genet. 2015, 96, 507–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Millan, F.; Cho, M.T.; Retterer, K.; Monaghan, K.G.; Bai, R.; Vitazka, P.; Everman, D.B.; Smith, B.; Angle, B.; Roberts, V.; et al. Whole exome sequencing reveals de novo pathogenic variants in KAT6A as a cause of a neurodevelopmental disorder. Am. J. Med. Genet. A 2016, 170, 1791–1798. [Google Scholar] [CrossRef] [PubMed]
- Fahrner, J.A.; Bjornsson, H.T. Mendelian disorders of the epigenetic machinery: Tipping the balance of chromatin states. Annu Rev. Genom. Hum. Genet. 2014, 15, 269–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avvakumov, N.; Cote, J. The MYST family of histone acetyltransferases and their intimate links to cancer. Oncogene 2007, 26, 5395–5407. [Google Scholar] [CrossRef] [Green Version]
- Voss, A.K.; Vanyai, H.K.; Collin, C.; Dixon, M.P.; McLennan, T.J.; Sheikh, B.N.; Scambler, P.; Thomas, T. MOZ regulates the Tbx1 locus, and Moz mutation partially phenocopies DiGeorge syndrome. Dev. Cell 2012, 23, 652–663. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.J. MOZ and MORF acetyltransferases: Molecular interaction, animal development and human disease. Biochim. Biophys. Acta 2015, 1853, 1818–1826. [Google Scholar] [CrossRef] [Green Version]
- Rozman, M.; Camos, M.; Colomer, D.; Villamor, N.; Esteve, J.; Costa, D.; Carrio, A.; Aymerich, M.; Aguilar, J.L.; Domingo, A.; et al. Type I MOZ/CBP (MYST3/CREBBP) is the most common chimeric transcript in acute myeloid leukemia with t(8;16)(p11;p13) translocation. Genes Chromosom. Cancer 2004, 40, 140–145. [Google Scholar] [CrossRef]
- Rokudai, S.; Laptenko, O.; Arnal, S.M.; Taya, Y.; Kitabayashi, I.; Prives, C. MOZ increases p53 acetylation and premature senescence through its complex formation with PML. Proc. Natl. Acad. Sci. USA 2013, 110, 3895–3900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelletier, N.; Champagne, N.; Stifani, S.; Yang, X.J. MOZ and MORF histone acetyltransferases interact with the Runt-domain transcription factor Runx2. Oncogene 2002, 21, 2729–2740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trinh, J.; Huning, I.; Yuksel, Z.; Baalmann, N.; Imhoff, S.; Klein, C.; Rolfs, A.; Gillessen-Kaesbach, G.; Lohmann, K. A KAT6A variant in a family with autosomal dominantly inherited microcephaly and developmental delay. J. Hum. Genet. 2018, 63, 997–1001. [Google Scholar] [CrossRef] [PubMed]
- Perez-Campo, F.M.; Borrow, J.; Kouskoff, V.; Lacaud, G. The histone acetyl transferase activity of monocytic leukemia zinc finger is critical for the proliferation of hematopoietic precursors. Blood 2009, 113, 4866–4874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sapountzi, V.; Cote, J. MYST-family histone acetyltransferases: Beyond chromatin. Cell. Mol. Life Sci. 2011, 68, 1147–1156. [Google Scholar] [CrossRef]
- Thomas, T.; Corcoran, L.M.; Gugasyan, R.; Dixon, M.P.; Brodnicki, T.; Nutt, S.L.; Metcalf, D.; Voss, A.K. Monocytic leukemia zinc finger protein is essential for the development of long-term reconstituting hematopoietic stem cells. Genes Dev. 2006, 20, 1175–1186. [Google Scholar] [CrossRef] [Green Version]
- Good-Jacobson, K.L.; Chen, Y.; Voss, A.K.; Smyth, G.K.; Thomas, T.; Tarlinton, D. Regulation of germinal center responses and B-cell memory by the chromatin modifier MOZ. Proc. Natl. Acad. Sci. USA 2014, 111, 9585–9590. [Google Scholar] [CrossRef] [Green Version]
- Newman, D.M.; Sakaguchi, S.; Lun, A.; Preston, S.; Pellegrini, M.; Khamina, K.; Bergthaler, A.; Nutt, S.L.; Smyth, G.K.; Voss, A.K.; et al. Acetylation of the Cd8 Locus by KAT6A Determines Memory T Cell Diversity. Cell Rep. 2016, 16, 3311–3321. [Google Scholar] [CrossRef] [Green Version]
- Villalon-Garcia, I.; Alvarez-Cordoba, M.; Suarez-Rivero, J.M.; Povea-Cabello, S.; Talaveron-Rey, M.; Suarez-Carrillo, A.; Munuera-Cabeza, M.; Sanchez-Alcazar, J.A. Precision Medicine in Rare Diseases. Diseases 2020, 8, 42. [Google Scholar] [CrossRef]
- Connolly, G.P. Fibroblast models of neurological disorders: Fluorescence measurement studies. Trends Pharm. Sci. 1998, 19, 171–177. [Google Scholar] [CrossRef]
- Gershlick, D.C.; Ishida, M.; Jones, J.R.; Bellomo, A.; Bonifacino, J.S.; Everman, D.B. A neurodevelopmental disorder caused by mutations in the VPS51 subunit of the GARP and EARP complexes. Hum. Mol. Genet. 2019, 28, 1548–1560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milev, M.P.; Graziano, C.; Karall, D.; Kuper, W.F.E.; Al-Deri, N.; Cordelli, D.M.; Haack, T.B.; Danhauser, K.; Iuso, A.; Palombo, F.; et al. Bi-allelic mutations in TRAPPC2L result in a neurodevelopmental disorder and have an impact on RAB11 in fibroblasts. J. Med. Genet. 2018, 55, 753–764. [Google Scholar] [CrossRef] [PubMed]
- Olesen, M.A.; Villavicencio-Tejo, F.; Quintanilla, R.A. The use of fibroblasts as a valuable strategy for studying mitochondrial impairment in neurological disorders. Transl. Neurodegener. 2022, 11, 36. [Google Scholar] [CrossRef] [PubMed]
- Sbardella, D.; Tundo, G.R.; Campagnolo, L.; Valacchi, G.; Orlandi, A.; Curatolo, P.; Borsellino, G.; D’Esposito, M.; Ciaccio, C.; Cesare, S.D.; et al. Retention of Mitochondria in Mature Human Red Blood Cells as the Result of Autophagy Impairment in Rett Syndrome. Sci. Rep. 2017, 7, 12297. [Google Scholar] [CrossRef] [Green Version]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 76, 7.20.1–7.20.41. [CrossRef] [Green Version]
- Alvarez-Cordoba, M.; Fernandez Khoury, A.; Villanueva-Paz, M.; Gomez-Navarro, C.; Villalon-Garcia, I.; Suarez-Rivero, J.M.; Povea-Cabello, S.; de la Mata, M.; Cotan, D.; Talaveron-Rey, M.; et al. Pantothenate Rescues Iron Accumulation in Pantothenate Kinase-Associated Neurodegeneration Depending on the Type of Mutation. Mol. Neurobiol. 2019, 56, 3638–3656. [Google Scholar] [CrossRef] [Green Version]
- Kamalian, L.; Douglas, O.; Jolly, C.E.; Snoeys, J.; Simic, D.; Monshouwer, M.; Williams, D.P.; Park, B.K.; Chadwick, A.E. Acute Metabolic Switch Assay Using Glucose/Galactose Medium in HepaRG Cells to Detect Mitochondrial Toxicity. Curr. Protoc. Toxicol. 2019, 80, e76. [Google Scholar] [CrossRef]
- Coelho, A.I.; Berry, G.T.; Rubio-Gozalbo, M.E. Galactose metabolism and health. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 422–427. [Google Scholar] [CrossRef]
- Rodriguez-Hernandez, A.; Cordero, M.D.; Salviati, L.; Artuch, R.; Pineda, M.; Briones, P.; Gomez Izquierdo, L.; Cotan, D.; Navas, P.; Sanchez-Alcazar, J.A. Coenzyme Q deficiency triggers mitochondria degradation by mitophagy. Autophagy 2009, 5, 19–32. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Alcalde, F.; Okonechnikov, K.; Carbonell, J.; Cruz, L.M.; Gotz, S.; Tarazona, S.; Dopazo, J.; Meyer, T.F.; Conesa, A. Qualimap: Evaluating next-generation sequencing alignment data. Bioinformatics 2012, 28, 2678–2679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Merico, D.; Isserlin, R.; Stueker, O.; Emili, A.; Bader, G.D. Enrichment map: A network-based method for gene-set enrichment visualization and interpretation. PLoS ONE 2010, 5, e13984. [Google Scholar] [CrossRef] [Green Version]
- Suarez-Rivero, J.M.; Pastor-Maldonado, C.J.; Romero-Gonzalez, A.; Gomez-Fernandez, D.; Povea-Cabello, S.; Alvarez-Cordoba, M.; Villalon-Garcia, I.; Talaveron-Rey, M.; Suarez-Carrillo, A.; Munuera-Cabeza, M.; et al. Pterostilbene in Combination With Mitochondrial Cofactors Improve Mitochondrial Function in Cellular Models of Mitochondrial Diseases. Front. Pharm. 2022, 13, 862085. [Google Scholar] [CrossRef]
- Suarez-Rivero, J.M.; Pastor-Maldonado, C.J.; Povea-Cabello, S.; Alvarez-Cordoba, M.; Villalon-Garcia, I.; Talaveron-Rey, M.; Suarez-Carrillo, A.; Munuera-Cabeza, M.; Reche-Lopez, D.; Cilleros-Holgado, P.; et al. UPR(mt) activation improves pathological alterations in cellular models of mitochondrial diseases. Orphanet J. Rare Dis. 2022, 17, 204. [Google Scholar] [CrossRef]
- Villanueva-Paz, M.; Povea-Cabello, S.; Villalon-Garcia, I.; Alvarez-Cordoba, M.; Suarez-Rivero, J.M.; Talaveron-Rey, M.; Jackson, S.; Falcon-Moya, R.; Rodriguez-Moreno, A.; Sanchez-Alcazar, J.A. Parkin-mediated mitophagy and autophagy flux disruption in cellular models of MERRF syndrome. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165726. [Google Scholar] [CrossRef]
- Le Boedec, K. Sensitivity and specificity of normality tests and consequences on reference interval accuracy at small sample size: A computer-simulation study. Vet. Clin. Pathol. 2016, 45, 648–656. [Google Scholar] [CrossRef]
- Alvarez-Cordoba, M.; Talaveron-Rey, M.; Villalon-Garcia, I.; Povea-Cabello, S.; Suarez-Rivero, J.M.; Suarez-Carrillo, A.; Munuera-Cabeza, M.; Salas, J.J.; Sanchez-Alcazar, J.A. Down regulation of the expression of mitochondrial phosphopantetheinyl-proteins in pantothenate kinase-associated neurodegeneration: Pathophysiological consequences and therapeutic perspectives. Orphanet J. Rare Dis. 2021, 16, 201. [Google Scholar] [CrossRef]
- Joshi, A.K.; Zhang, L.; Rangan, V.S.; Smith, S. Cloning, expression, and characterization of a human 4′-phosphopantetheinyl transferase with broad substrate specificity. J. Biol. Chem. 2003, 278, 33142–33149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoshan-Barmatz, V.; Ben-Hail, D. VDAC, a multi-functional mitochondrial protein as a pharmacological target. Mitochondrion 2012, 12, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.J. The diverse superfamily of lysine acetyltransferases and their roles in leukemia and other diseases. Nucleic Acids Res. 2004, 32, 959–976. [Google Scholar] [CrossRef] [Green Version]
- Berger, S.L. Histone modifications in transcriptional regulation. Curr. Opin. Genet. Dev. 2002, 12, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Glass, C.K.; Rosenfeld, M.G. The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev. 2000, 14, 121–141. [Google Scholar] [CrossRef]
- Taatjes, D.J.; Marr, M.T.; Tjian, R. Regulatory diversity among metazoan co-activator complexes. Nat. Rev. Mol. Cell Biol. 2004, 5, 403–410. [Google Scholar] [CrossRef]
- Howe, L.; Auston, D.; Grant, P.; John, S.; Cook, R.G.; Workman, J.L.; Pillus, L. Histone H3 specific acetyltransferases are essential for cell cycle progression. Genes Dev. 2001, 15, 3144–3154. [Google Scholar] [CrossRef] [Green Version]
- Katsumoto, T.; Aikawa, Y.; Iwama, A.; Ueda, S.; Ichikawa, H.; Ochiya, T.; Kitabayashi, I. MOZ is essential for maintenance of hematopoietic stem cells. Genes Dev. 2006, 20, 1321–1330. [Google Scholar] [CrossRef] [Green Version]
- Sheikh, B.N.; Lee, S.C.; El-Saafin, F.; Vanyai, H.K.; Hu, Y.; Pang, S.H.; Grabow, S.; Strasser, A.; Nutt, S.L.; Alexander, W.S.; et al. MOZ regulates B-cell progenitors and, consequently, Moz haploinsufficiency dramatically retards MYC-induced lymphoma development. Blood 2015, 125, 1910–1921. [Google Scholar] [CrossRef]
- Huang, F.; Abmayr, S.M.; Workman, J.L. Regulation of KAT6 Acetyltransferases and Their Roles in Cell Cycle Progression, Stem Cell Maintenance, and Human Disease. Mol. Cell. Biol. 2016, 36, 1900–1907. [Google Scholar] [CrossRef]
- Sheikh, B.N.; Phipson, B.; El-Saafin, F.; Vanyai, H.K.; Downer, N.L.; Bird, M.J.; Kueh, A.J.; May, R.E.; Smyth, G.K.; Voss, A.K.; et al. MOZ (MYST3, KAT6A) inhibits senescence via the INK4A-ARF pathway. Oncogene 2015, 34, 5807–5820. [Google Scholar] [CrossRef] [PubMed]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Eberharter, A.; Becker, P.B. Histone acetylation: A switch between repressive and permissive chromatin. Second in review series on chromatin dynamics. EMBO Rep. 2002, 3, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Voss, A.K.; Collin, C.; Dixon, M.P.; Thomas, T. Moz and retinoic acid coordinately regulate H3K9 acetylation, Hox gene expression, and segment identity. Dev. Cell 2009, 17, 674–686. [Google Scholar] [CrossRef] [Green Version]
- Kitabayashi, I.; Aikawa, Y.; Nguyen, L.A.; Yokoyama, A.; Ohki, M. Activation of AML1-mediated transcription by MOZ and inhibition by the MOZ-CBP fusion protein. EMBO J. 2001, 20, 7184–7196. [Google Scholar] [CrossRef] [Green Version]
- Bristow, C.A.; Shore, P. Transcriptional regulation of the human MIP-1alpha promoter by RUNX1 and MOZ. Nucleic Acids Res. 2003, 31, 2735–2744. [Google Scholar] [CrossRef] [Green Version]
- Hiltunen, J.K.; Schonauer, M.S.; Autio, K.J.; Mittelmeier, T.M.; Kastaniotis, A.J.; Dieckmann, C.L. Mitochondrial fatty acid synthesis type II: More than just fatty acids. J. Biol. Chem. 2009, 284, 9011–9015. [Google Scholar] [CrossRef] [Green Version]
- Cronan, J.E. Assembly of Lipoic Acid on Its Cognate Enzymes: An Extraordinary and Essential Biosynthetic Pathway. Microbiol. Mol. Biol. Rev. 2016, 80, 429–450. [Google Scholar] [CrossRef] [Green Version]
- Millichap, L.E.; Damiani, E.; Tiano, L.; Hargreaves, I.P. Targetable Pathways for Alleviating Mitochondrial Dysfunction in Neurodegeneration of Metabolic and Non-Metabolic Diseases. Int. J. Mol. Sci. 2021, 22, 11444. [Google Scholar] [CrossRef]
- Mignani, L.; Gnutti, B.; Zizioli, D.; Finazzi, D. Coenzyme a Biochemistry: From Neurodevelopment to Neurodegeneration. Brain Sci. 2021, 11, 1031. [Google Scholar] [CrossRef]
- Tahiliani, A.G.; Beinlich, C.J. Pantothenic acid in health and disease. Vitam. Horm. 1991, 46, 165–228. [Google Scholar] [CrossRef] [PubMed]
- Rock, C.O.; Calder, R.B.; Karim, M.A.; Jackowski, S. Pantothenate kinase regulation of the intracellular concentration of coenzyme A. J. Biol. Chem. 2000, 275, 1377–1383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bremer, J. Carnitine—metabolism and functions. Physiol. Rev. 1983, 63, 1420–1480. [Google Scholar] [CrossRef] [PubMed]
- Jacob, C.; Belleville, F. L-carnitine: Metabolism, functions and value in pathology. Pathol.-Biol. 1992, 40, 910–919. [Google Scholar]
- Huang, H.; Liu, N.; Guo, H.; Liao, S.; Li, X.; Yang, C.; Liu, S.; Song, W.; Liu, C.; Guan, L.; et al. L-carnitine is an endogenous HDAC inhibitor selectively inhibiting cancer cell growth in vivo and in vitro. PLoS ONE 2012, 7, e49062. [Google Scholar] [CrossRef] [Green Version]
- McCarty, M.; DiNicolantonio, J.J.; O’Keefe, J.H. The Ability of Carnitine to Act as a Type 1Histone Deacetylase Inhibitor May Explain the Favorable Impact of Carnitine Supplementation on Mitochondrial Biogenesis in the Elderly. Med. Res. Arch. 2020, 8. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Munuera-Cabeza, M.; Álvarez-Córdoba, M.; Suárez-Rivero, J.M.; Povea-Cabello, S.; Villalón-García, I.; Talaverón-Rey, M.; Suárez-Carrillo, A.; Reche-López, D.; Cilleros-Holgado, P.; Piñero-Pérez, R.; et al. Pantothenate and L-Carnitine Supplementation Improves Pathological Alterations in Cellular Models of KAT6A Syndrome. Genes 2022, 13, 2300. https://doi.org/10.3390/genes13122300
Munuera-Cabeza M, Álvarez-Córdoba M, Suárez-Rivero JM, Povea-Cabello S, Villalón-García I, Talaverón-Rey M, Suárez-Carrillo A, Reche-López D, Cilleros-Holgado P, Piñero-Pérez R, et al. Pantothenate and L-Carnitine Supplementation Improves Pathological Alterations in Cellular Models of KAT6A Syndrome. Genes. 2022; 13(12):2300. https://doi.org/10.3390/genes13122300
Chicago/Turabian StyleMunuera-Cabeza, Manuel, Mónica Álvarez-Córdoba, Juan M. Suárez-Rivero, Suleva Povea-Cabello, Irene Villalón-García, Marta Talaverón-Rey, Alejandra Suárez-Carrillo, Diana Reche-López, Paula Cilleros-Holgado, Rocío Piñero-Pérez, and et al. 2022. "Pantothenate and L-Carnitine Supplementation Improves Pathological Alterations in Cellular Models of KAT6A Syndrome" Genes 13, no. 12: 2300. https://doi.org/10.3390/genes13122300