
An Efficient Feature Selection Algorithm for Gene Families Using NMF and ReliefF

Abstract
:1. Introduction
2. Materials and Methods
2.1. Genome Resources and Species Selection
2.2. Gene Family Analysis
2.3. Feature Selection

2.4. Classification Methods
2.5. Prediction Accuracy Assessment
3. Results and Discussion
3.1. Data Sets
3.2. The Selection of Classifier
3.3. Classifying Insect Feeding Habits by Machine Learning
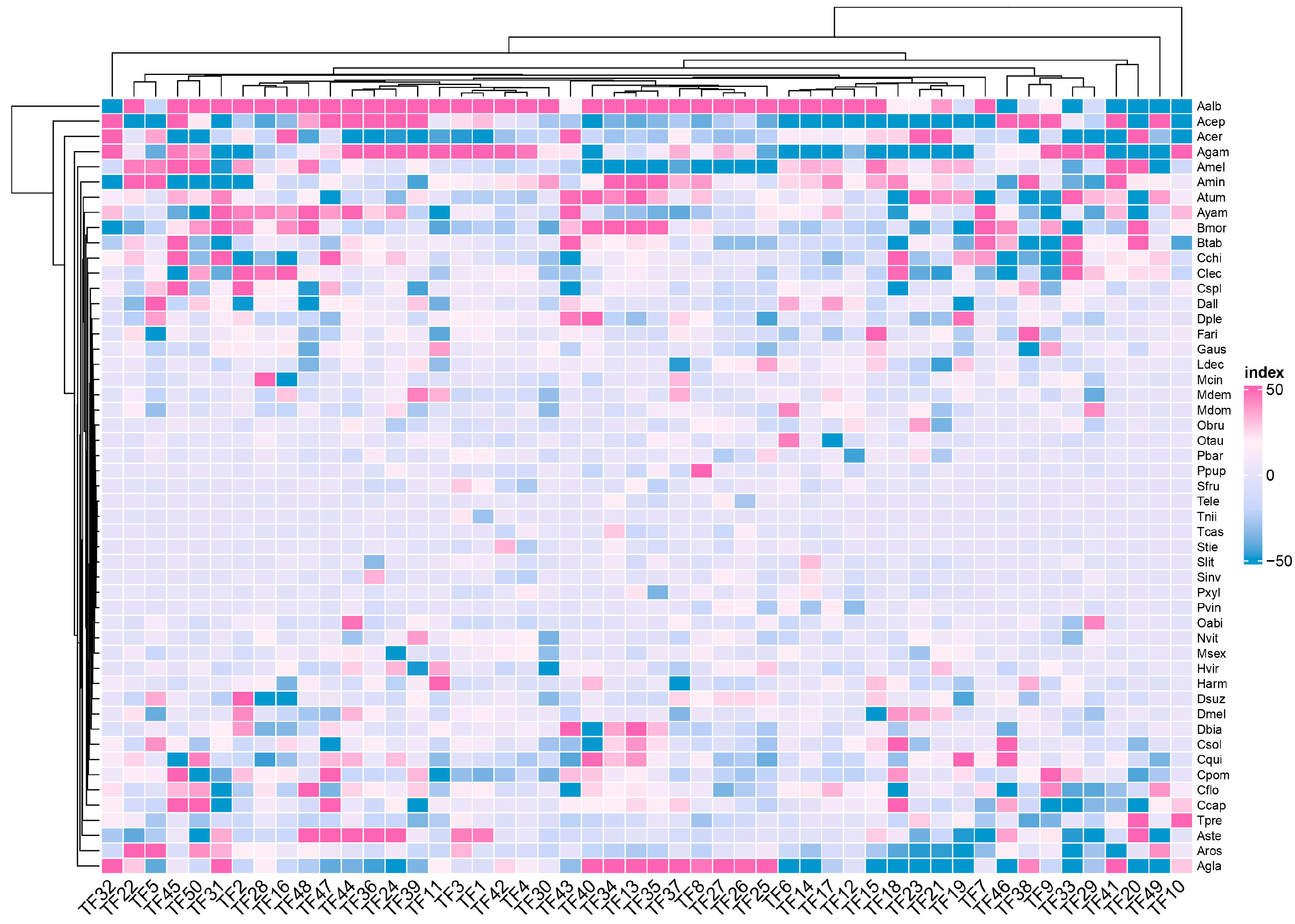
3.4. Feature Selected Reflect the Relationship of Gene Family
3.5. Comparison with Other Gene Selection Methods
3.6. The Relationships of Selected Features
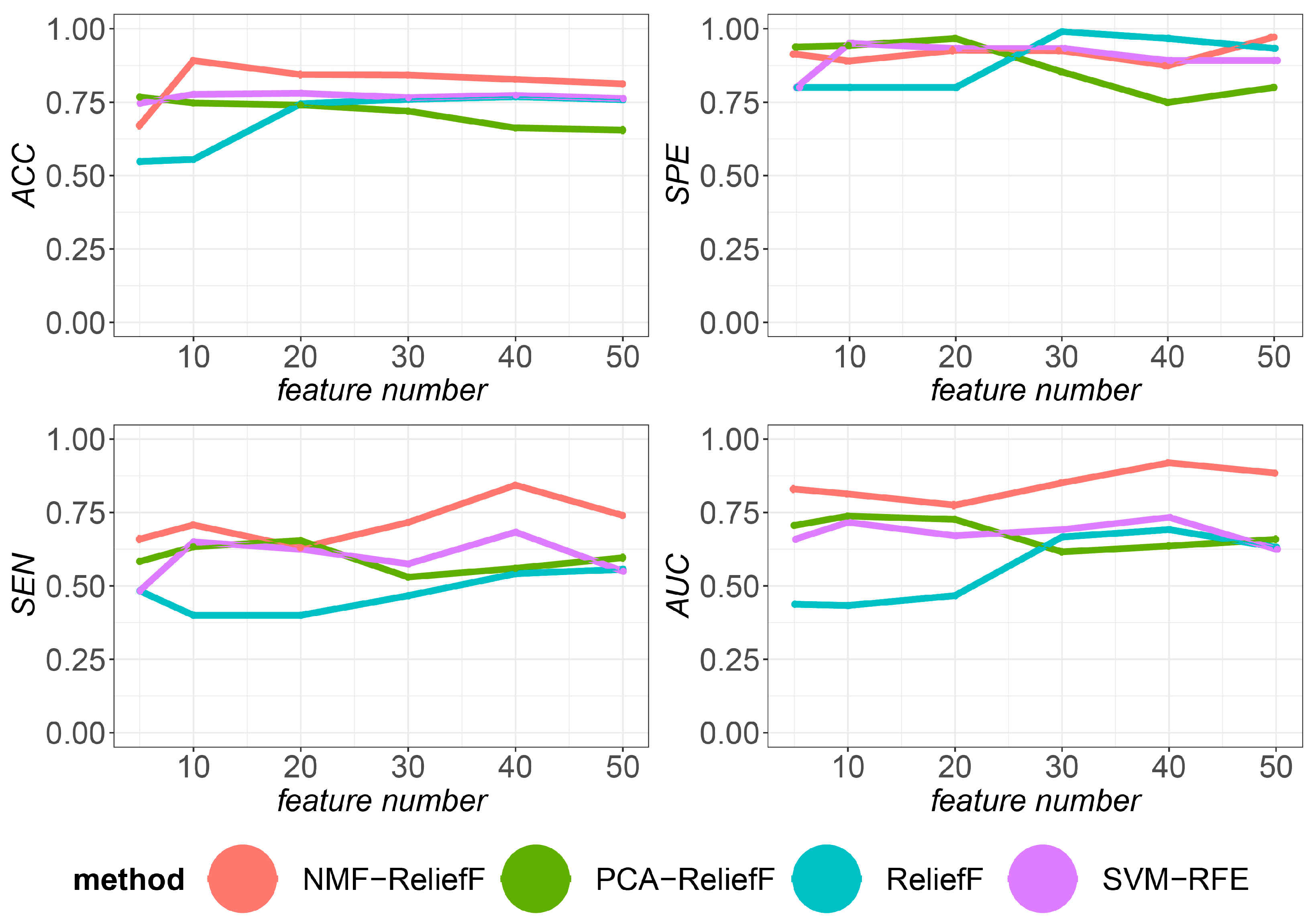
3.7. Classification Performance with Different Numbers of Selected Gene Families
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Demuth, J.P.; Bie, T.D.; Stajich, J.E.; Cristianini, N.; Hahn, M.W. The Evolution of Mammalian Gene Families. PLoS ONE 2006, 1, e85. [Google Scholar] [CrossRef] [PubMed]
- Liberles, D.A.; Dittmar, K. Characterizing Gene Family Evolution. Biol. Proced. Online 2008, 10, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Hartwell, L.H.; Hood, L.; Goldberg, M.L.; Reynolds, A.E.; Silver, L.M. Genetics from Genes to Genomes, 4th ed.; McGraw-Hill: New York, NY, USA, 2011. [Google Scholar]
- Luna, S.K.; Chain, F.J.J. Lineage-Specific Genes and Family Expansions in Dictyostelid Genomes Display Expression Bias and Evolutionary Diversification during Development. Genes 2021, 12, 1628. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.-C.; Guo, Y.-L. Less Is More, Natural Loss-of-Function Mutation Is a Strategy for Adaptation. Plant Commun. 2020, 1, 100103. [Google Scholar] [CrossRef]
- Demuth, J.P.; Hahn, M.W. The Life and Death of Gene Families. Bioessays 2009, 31, 29–39. [Google Scholar] [CrossRef]
- Panfilio, K.A.; Vargas Jentzsch, I.M.; Benoit, J.B.; Erezyilmaz, D.; Suzuki, Y.; Colella, S.; Robertson, H.M.; Poelchau, M.F.; Waterhouse, R.M.; Ioannidis, P.; et al. Molecular Evolutionary Trends and Feeding Ecology Diversification in the Hemiptera, Anchored by the Milkweed Bug Genome. Genome Biol. 2019, 20, 64. [Google Scholar] [CrossRef]
- Xu, H.; Zhao, X.; Yang, Y.; Chen, X.; Mei, Y.; He, K.; Xu, L.; Ye, X.; Liu, Y.; Li, F.; et al. Chromosome-Level Genome Assembly of an Agricultural Pest, the Rice Leaffolder Cnaphalocrocis Exigua (Crambidae, Lepidoptera). Mol. Ecol. Resour. 2022, 22, 307–318. [Google Scholar] [CrossRef]
- Zheng, X.; Zhu, Q.; Zhou, Z.; Wu, F.; Chen, L.; Cao, Q. Gut Bacterial Communities across 12 Ensifera (Orthoptera) at Different Feeding Habits and Its Prediction for the Insect with Contrasting Feeding Habits. PLoS ONE 2021, 16, e0250675. [Google Scholar] [CrossRef]
- Lippert, C.; Sabatini, R.; Maher, M.C.; Kang, E.Y.; Lee, S.; Arikan, O.; Harley, A.; Bernal, A.; Garst, P.; Lavrenko, V.; et al. Identification of Individuals by Trait Prediction Using Whole-Genome Sequencing Data. Proc. Natl. Acad. Sci. USA 2017, 114, 10166–10171. [Google Scholar] [CrossRef]
- Alexeyenko, A.; Lindberg, J.; Pérez-Bercoff, Å.; Sonnhammer, E.L.L. Overview and Comparison of Ortholog Databases. Drug Discov. Today Technol. 2006, 3, 137–143. [Google Scholar] [CrossRef]
- Altenhoff, A.M.; Train, C.-M.; Gilbert, K.J.; Mediratta, I.; Mendes de Farias, T.; Moi, D.; Nevers, Y.; Radoykova, H.-S.; Rossier, V.; Warwick Vesztrocy, A.; et al. OMA Orthology in 2021: Website Overhaul, Conserved Isoforms, Ancestral Gene Order and More. Nucleic Acids Res. 2020, 49, D373–D379. [Google Scholar] [CrossRef]
- Zdobnov, E.M.; Kuznetsov, D.; Tegenfeldt, F.; Manni, M.; Berkeley, M.; Kriventseva, E.V. OrthoDB in 2020: Evolutionary and Functional Annotations of Orthologs. Nucleic Acids Res. 2021, 49, D389–D393. [Google Scholar] [CrossRef]
- Schreiber, F.; Patricio, M.; Muffato, M.; Pignatelli, M.; Bateman, A. TreeFam v9: A New Website, More Species and Orthology-on-the-Fly. Nucleic Acids Res. 2014, 42, D922–D925. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J.; et al. EggNOG 5.0: A Hierarchical, Functionally and Phylogenetically Annotated Orthology Resource Based on 5090 Organisms and 2502 Viruses. Nucleic Acids Res. 2019, 47, D309–D314. [Google Scholar] [CrossRef]
- Kristensen, D.M.; Wolf, Y.I.; Mushegian, A.R.; Koonin, E.V. Computational Methods for Gene Orthology Inference. Brief. Bioinform. 2011, 12, 379–391. [Google Scholar] [CrossRef]
- Trachana, K.; Larsson, T.A.; Powell, S.; Chen, W.-H.; Doerks, T.; Muller, J.; Bork, P. Orthology Prediction Methods: A Quality Assessment Using Curated Protein Families. Bioessays 2011, 33, 769–780. [Google Scholar] [CrossRef]
- The UniProt Consortium. UniProt: The Universal Protein Knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar] [CrossRef]
- Mirjalili, S. Evolutionary Algorithms and Neural Networks. In Studies in Computational Intelligence; Springer International Publishing: Cham, Germany, 2019; Volume 780, ISBN 978-3-319-93024-4. [Google Scholar]
- Qi, Y. Random Forest for Bioinformatics. In Ensemble Machine Learning: Methods and Applications; Zhang, C., Ma, Y., Eds.; Springer: Boston, MA, USA, 2012; pp. 307–323. ISBN 978-1-4419-9326-7. [Google Scholar]
- Xie, J.; Girshick, R.; Farhadi, A. Unsupervised Deep Embedding for Clustering Analysis. In Proceedings of the 33rd International Conference on Machine Learning, New York, NY, USA, 19–24 June 2016; pp. 478–487. [Google Scholar]
- Urbanowicz, R.J.; Meeker, M.; La Cava, W.; Olson, R.S.; Moore, J.H. Relief-Based Feature Selection: Introduction and Review. J. Biomed. Inform. 2018, 85, 189–203. [Google Scholar] [CrossRef]
- Abdi, H.; Williams, L.J. Principal Component Analysis. WIREs Comput. Stat. 2010, 2, 433–459. [Google Scholar] [CrossRef]
- Spolaôr, N.; Cherman, E.A.; Monard, M.C.; Lee, H.D. ReliefF for Multi-Label Feature Selection. In Proceedings of the 2013 Brazilian Conference on Intelligent Systems, Fortaleza, Brazil, 19–24 October 2013; pp. 6–11. [Google Scholar]
- Chen, Q.; Meng, Z.; Liu, X.; Jin, Q.; Su, R. Decision Variants for the Automatic Determination of Optimal Feature Subset in RF-RFE. Genes 2018, 9, 301. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Jiang, X.; Xu, J.; Quan, X.; Wu, M.; Zhang, H. Ensemble Consensus-Guided Unsupervised Feature Selection to Identify Huntington’s Disease-Associated Genes. Genes 2018, 9, 350. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Zhang, H.; Zhang, Z.; Quan, X. Flexible Non-Negative Matrix Factorization to Unravel Disease-Related Genes. IEEE/ACM Trans. Comput. Biol. Bioinform. 2019, 16, 1948–1957. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Sidiropoulos, N.D.; Swami, A. Non-Negative Matrix Factorization Revisited: Uniqueness and Algorithm for Symmetric Decomposition. IEEE Trans. Signal Process. 2014, 62, 211–224. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, B.; Fang, Y. Evolution of Insect Diversity in the Jehol Biota. Sci. China Earth Sci. 2010, 53, 1908–1917. [Google Scholar] [CrossRef]
- Li, F.; Zhao, X.; Li, M.; He, K.; Huang, C.; Zhou, Y.; Li, Z.; Walters, J.R. Insect Genomes: Progress and Challenges. Insect Mol. Biol. 2019, 28, 739–758. [Google Scholar] [CrossRef]
- Pruitt, K.D.; Tatusova, T.; Maglott, D.R. NCBI Reference Sequence (RefSeq): A Curated Non-Redundant Sequence Database of Genomes, Transcripts and Proteins. Nucleic Acids Res. 2005, 33, D501–D504. [Google Scholar] [CrossRef]
- Mei, Y.; Jing, D.; Tang, S.; Chen, X.; Chen, H.; Duanmu, H.; Cong, Y.; Chen, M.; Ye, X.; Zhou, H.; et al. InsectBase 2.0: A Comprehensive Gene Resource for Insects. Nucleic Acids Res. 2022, 50, D1040–D1045. [Google Scholar] [CrossRef]
- Amos, B.; Aurrecoechea, C.; Barba, M.; Barreto, A.; Basenko, E.Y.; Bażant, W.; Belnap, R.; Blevins, A.S.; Böhme, U.; Brestelli, J.; et al. VEuPathDB: The Eukaryotic Pathogen, Vector and Host Bioinformatics Resource Center. Nucleic Acids Res. 2022, 50, D898–D911. [Google Scholar] [CrossRef]
- Fallon, T.R.; Lower, S.E.; Chang, C.-H.; Bessho-Uehara, M.; Martin, G.J.; Bewick, A.J.; Behringer, M.; Debat, H.J.; Wong, I.; Day, J.C.; et al. Firefly Genomes Illuminate Parallel Origins of Bioluminescence in Beetles. eLife 2018, 7, e36495. [Google Scholar] [CrossRef]
- Yates, A.D.; Allen, J.; Amode, R.M.; Azov, A.G.; Barba, M.; Becerra, A.; Bhai, J.; Campbell, L.I.; Carbajo Martinez, M.; Chakiachvili, M.; et al. Ensembl Genomes 2022: An Expanding Genome Resource for Non-Vertebrates. Nucleic Acids Res. 2022, 50, D996–D1003. [Google Scholar] [CrossRef]
- Sneddon, T.P.; Li, P.; Edmunds, S.C. GigaDB: Announcing the GigaScience Database. Gigascience 2012, 1, 11. [Google Scholar] [CrossRef]
- Mundra, P.A.; Rajapakse, J.C. SVM-RFE With MRMR Filter for Gene Selection. IEEE Transactions on NanoBioscience 2010, 9, 31–37. [Google Scholar] [CrossRef]
- Kira, K.; Rendell, L.A. The Feature Selection Problem: Traditional Methods and a New Algorithm. In Proceedings of the AAAI, San Jose, CA, USA, 12–16 July 1992; Volume 2, pp. 129–134. [Google Scholar]
- Zeng, X.; Wang, Q.; Zhang, C.; Cai, H. Feature Selection Based on ReliefF and PCA for Underwater Sound Classification. In Proceedings of the Proceedings of 2013 3rd International Conference on Computer Science and Network Technology, Dalian, China, 12–13 October 2013; pp. 442–445. [Google Scholar]
- Zoidi, O.; Tefas, A.; Pitas, I. Multiplicative Update Rules for Concurrent Nonnegative Matrix Factorization and Maximum Margin Classification. IEEE Trans. Neural. Netw. Learn. Syst. 2013, 24, 422–434. [Google Scholar] [CrossRef]
- Le, T.T.; Urbanowicz, R.J.; Moore, J.H.; McKinney, B.A. STatistical Inference Relief (STIR) Feature Selection. Bioinformatics 2019, 35, 1358–1365. [Google Scholar] [CrossRef]
- Byvatov, E.; Schneider, G. Support Vector Machine Applications in Bioinformatics. Appl. Bioinform. 2003, 2, 67–77. [Google Scholar]
- Jiang, L.; Cai, Z.; Wang, D.; Jiang, S. Survey of Improving K-Nearest-Neighbor for Classification. In Proceedings of the Fourth International Conference on Fuzzy Systems and Knowledge Discovery (FSKD 2007), Haikou, China, 24–27 August 2007; Volume 1, pp. 679–683. [Google Scholar]
- Fushiki, T. Estimation of Prediction Error by Using K-Fold Cross-Validation. Stat. Comp. 2011, 21, 137–146. [Google Scholar] [CrossRef]
- Bolón-Canedo, V.; Sánchez-Maroño, N.; Alonso-Betanzos, A.; Benítez, J.M.; Herrera, F. A Review of Microarray Datasets and Applied Feature Selection Methods. Inf. Sci. 2014, 282, 111–135. [Google Scholar] [CrossRef]
- Cilia, N.D.; De Stefano, C.; Fontanella, F.; Raimondo, S.; Scotto di Freca, A. An Experimental Comparison of Feature-Selection and Classification Methods for Microarray Datasets. Information 2019, 10, 109. [Google Scholar] [CrossRef]
- Remeseiro, B.; Bolon-Canedo, V. A Review of Feature Selection Methods in Medical Applications. Comput. Biol. Med. 2019, 112, 103375. [Google Scholar] [CrossRef]
- Gordon, G.J.; Jensen, R.V.; Hsiao, L.-L.; Gullans, S.R.; Blumenstock, J.E.; Ramaswamy, S.; Richards, W.G.; Sugarbaker, D.J.; Bueno, R. Translation of Microarray Data into Clinically Relevant Cancer Diagnostic Tests Using Gene Expression Ratios in Lung Cancer and Mesothelioma. Cancer Res. 2002, 62, 4963–4967. [Google Scholar]
- Tian, E.; Zhan, F.; Walker, R.; Rasmussen, E.; Ma, Y.; Barlogie, B.; Shaughnessy, J.D. The Role of the Wnt-Signaling Antagonist DKK1 in the Development of Osteolytic Lesions in Multiple Myeloma. N. Engl. J. Med. 2003, 349, 2483–2494. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.; Febbo, P.G.; Ross, K.; Jackson, D.G.; Manola, J.; Ladd, C.; Tamayo, P.; Renshaw, A.A.; D’Amico, A.V.; Richie, J.P.; et al. Gene Expression Correlates of Clinical Prostate Cancer Behavior. Cancer Cell 2002, 1, 203–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, M.; Blanchette, C.; Dressman, H.; Huang, E.; Ishida, S.; Spang, R.; Zuzan, H.; Olson, J.A.; Marks, J.R.; Nevins, J.R. Predicting the Clinical Status of Human Breast Cancer by Using Gene Expression Profiles. Proc. Natl. Acad. Sci. USA 2001, 98, 11462–11467. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Sets | Instance | Gene Number | Class | Disease |
|---|---|---|---|---|
| Gordon [48] | 181 | 12,533 | 2 | Lung Cancer |
| Tian [49] | 173 | 12,625 | 2 | Myeloma |
| Singh [50] | 102 | 12,600 | 2 | Prostate Cancer |
| West [51] | 49 | 7129 | 2 | Breast Cancer |
| Lung | Prostate | Myeloma | Breast | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Methods | Classifiers | ACC | SEN | SPE | AUC | ACC | SEN | SPE | AUC | ACC | SEN | SPE | AUC | ACC | SEN | SPN | AUC |
| ReliefF | k-NN | 0.857 | 0.990 | 0.714 | 0.866 | 0.804 | 0.472 | 0.896 | 0.950 | 0.803 | 0.788 | 0.849 | 0.667 | 0.914 | 0.833 | 0.996 | 0.833 |
| RF | 0.757 | 0.658 | 0.859 | 0.794 | 0.798 | 0.320 | 0.928 | 0.952 | 0.843 | 0.876 | 0.836 | 0.739 | 0.767 | 0.553 | 0.910 | 0.460 | |
| SVM | 0.847 | 0.864 | 0.858 | 0.880 | 0.809 | 0.129 | 0.985 | 0.988 | 0.883 | 0.888 | 0.904 | 0.801 | 0.904 | 0.900 | 0.967 | 0.867 | |
| SVM-RFE | k-NN | 0.757 | 0.871 | 0.643 | 0.829 | 0.758 | 0.871 | 0.643 | 0.830 | 0.764 | 0.720 | 0.809 | 0.082 | 0.852 | 0.678 | 0.997 | 0.668 |
| RF | 0.815 | 0.810 | 0.810 | 0.847 | 0.816 | 0.810 | 0.810 | 0.847 | 0.775 | 0.810 | 0.751 | 0.602 | 0.791 | 0.667 | 0.883 | 0.589 | |
| SVM | 0.847 | 0.860 | 0.883 | 0.849 | 0.848 | 0.860 | 0.883 | 0.850 | 0.892 | 0.880 | 0.906 | 0.799 | 0.910 | 0.867 | 0.950 | 0.833 | |
| PCA-ReliefF | k-NN | 0.573 | 0.740 | 0.370 | 0.645 | 0.774 | 0.475 | 0.847 | 0.918 | 0.774 | 0.475 | 0.847 | 0.918 | 0.652 | 0.300 | 0.800 | 0.220 |
| RF | 0.531 | 0.540 | 0.566 | 0.639 | 0.769 | 0.239 | 0.898 | 0.927 | 0.769 | 0.239 | 0.898 | 0.927 | 0.848 | 0.920 | 0.880 | 0.800 | |
| SVM | 0.546 | 0.560 | 0.550 | 0.602 | 0.878 | 0.581 | 0.956 | 0.980 | 0.878 | 0.581 | 0.956 | 0.980 | 0.850 | 0.960 | 0.927 | 0.807 | |
| NMF-ReliefF | k-NN | 0.751 | 0.943 | 0.914 | 0.709 | 0.919 | 0.943 | 0.986 | 0.998 | 0.921 | 0.948 | 0.966 | 0.845 | 0.848 | 0.948 | 0.833 | 0.700 |
| RF | 0.593 | 0.567 | 0.657 | 0.619 | 0.873 | 0.673 | 0.933 | 0.980 | 0.940 | 0.946 | 0.940 | 0.891 | 0.881 | 0.800 | 0.883 | 0.750 | |
| SVM | 0.855 | 0.846 | 0.902 | 0.843 | 0.942 | 0.833 | 0.978 | 0.985 | 0.941 | 0.983 | 0.915 | 0.898 | 0.914 | 0.800 | 0.933 | 0.800 | |
| Baseline | k-NN | 0.545 | 0.783 | 0.425 | 0.550 | 0.734 | 0.325 | 0.836 | 0.883 | 0.682 | 0.688 | 0.739 | 0.509 | 0.557 | 0.400 | 0.653 | 0.317 |
| RF | 0.545 | 0.575 | 0.508 | 0.600 | 0.774 | 0.233 | 0.931 | 0.956 | 0.717 | 0.783 | 0.713 | 0.553 | 0.467 | 0.417 | 0.503 | 0.200 | |
| SVM | 0.575 | 0.558 | 0.475 | 0.583 | 0.687 | 0.252 | 0.803 | 0.854 | 0.872 | 0.912 | 0.859 | 0.788 | 0.710 | 0.750 | 0.667 | 0.717 | |
| Methods | Classifers | ACC | SEN | SPE | AUC | Time |
|---|---|---|---|---|---|---|
| ReliefF | k-NN | 0.766 | 0.758 | 0.833 | 0.700 | 1.063 |
| RF | 0.783 | 0.675 | 0.916 | 0.750 | 1.756 | |
| SVM | 0.786 | 0.541 | 0.966 | 0.691 | 1.074 | |
| SVM-RFE | k-NN | 0.770 | 0.708 | 0.866 | 0.725 | 3.396 |
| RF | 0.730 | 0.675 | 0.825 | 0.708 | 4.057 | |
| SVM | 0.786 | 0.683 | 0.891 | 0.733 | 3.492 | |
| PCA-ReliefF | k-NN | 0.669 | 0.573 | 0.749 | 0.653 | 0.060 |
| RF | 0.609 | 0.526 | 0.609 | 0.609 | 0.644 | |
| SVM | 0.667 | 0.560 | 0.744 | 0.636 | 0.066 | |
| NMF-ReliefF | k-NN | 0.745 | 0.443 | 0.370 | 0.788 | 1.310 |
| RF | 0.723 | 0.696 | 0.765 | 0.800 | 2.038 | |
| SVM | 0.843 | 0.843 | 0.974 | 0.919 | 1.324 | |
| Baseline | k-NN | 0.643 | 0.750 | 0.725 | 0.629 | 0.069 |
| RF | 0.663 | 0.600 | 0.650 | 0.587 | 9.657 | |
| SVM | 0.683 | 0.566 | 0.783 | 0.629 | 0.097 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, K.; Chen, Q.; Huang, G.-H. An Efficient Feature Selection Algorithm for Gene Families Using NMF and ReliefF. Genes 2023, 14, 421. https://doi.org/10.3390/genes14020421
Liu K, Chen Q, Huang G-H. An Efficient Feature Selection Algorithm for Gene Families Using NMF and ReliefF. Genes. 2023; 14(2):421. https://doi.org/10.3390/genes14020421
Chicago/Turabian StyleLiu, Kai, Qi Chen, and Guo-Hua Huang. 2023. "An Efficient Feature Selection Algorithm for Gene Families Using NMF and ReliefF" Genes 14, no. 2: 421. https://doi.org/10.3390/genes14020421