



Genes 2024, 15(4), 449; https://doi.org/10.3390/genes15040449 - 02 Apr 2024
Viewed by 489
Abstract
►
Show Figures
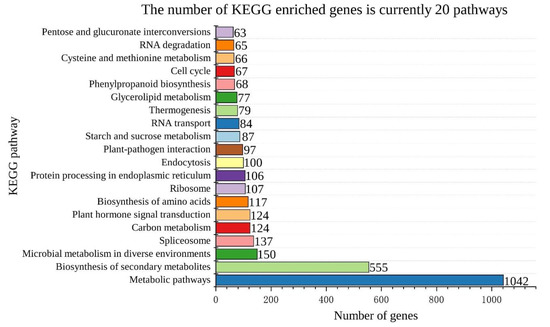
Background: Alfalfa, the most economically important forage legume worldwide, features modest genetic progress due to long selection cycles and the extent of the non-additive genetic variance associated with its autotetraploid genome. Methods: To improve the efficiency of genomic selection in alfalfa, we explored
[...] Read more.
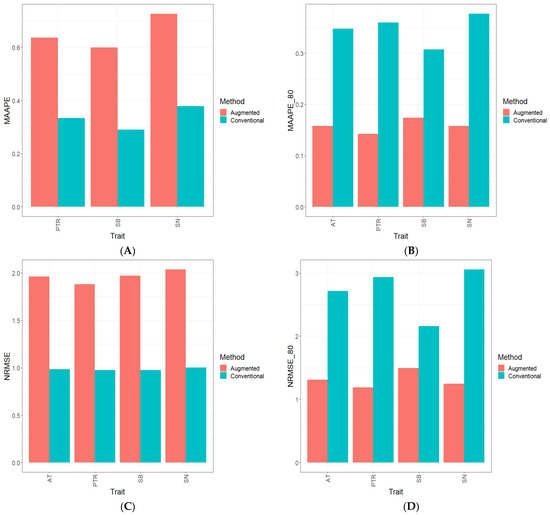
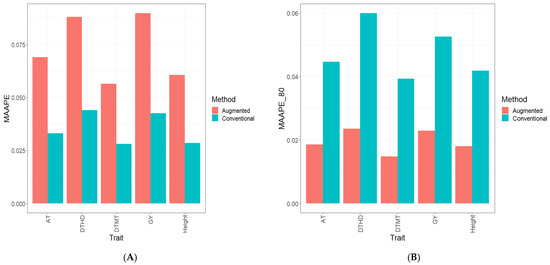
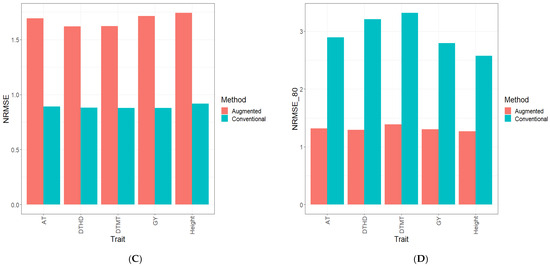
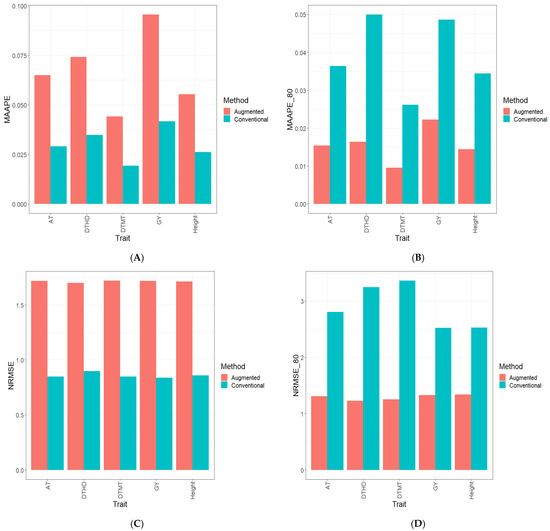
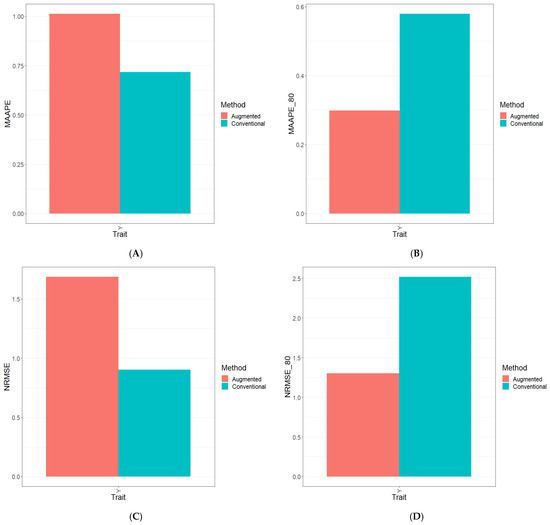
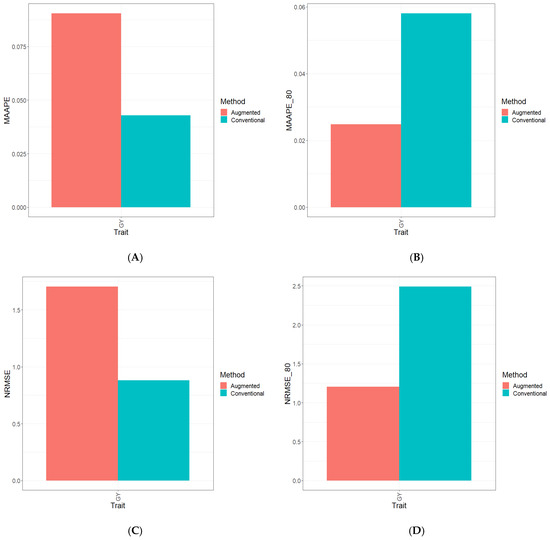
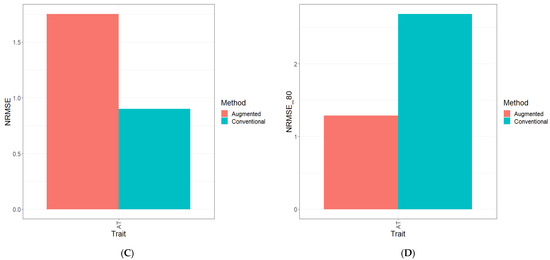
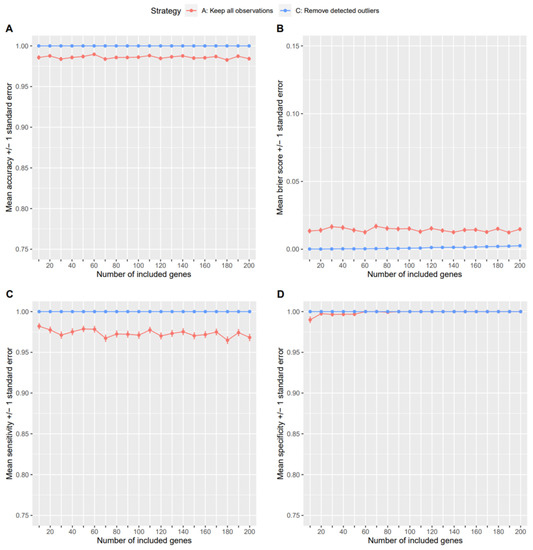
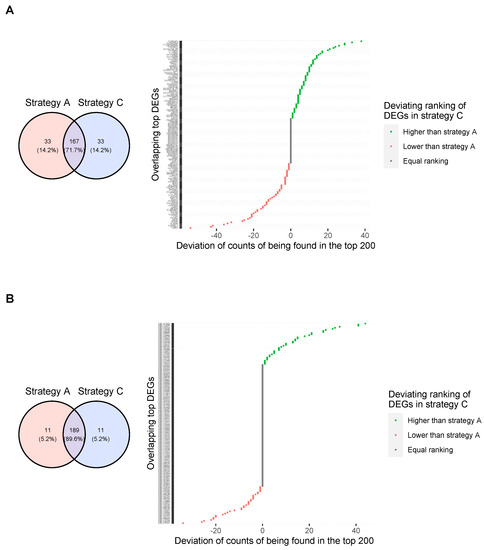
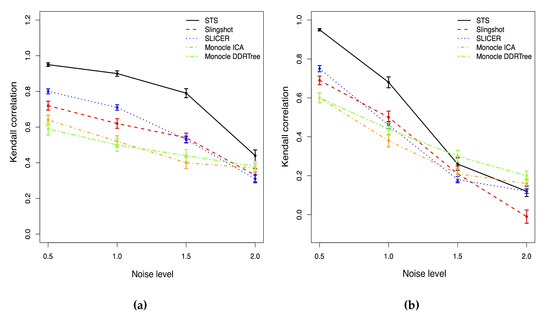
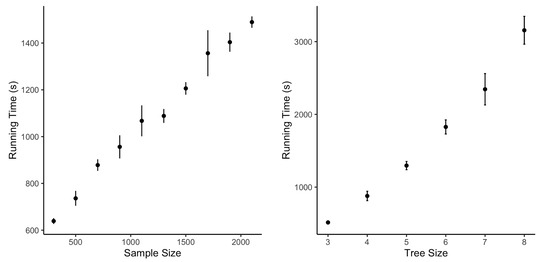
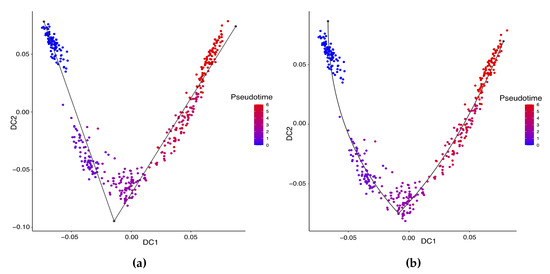
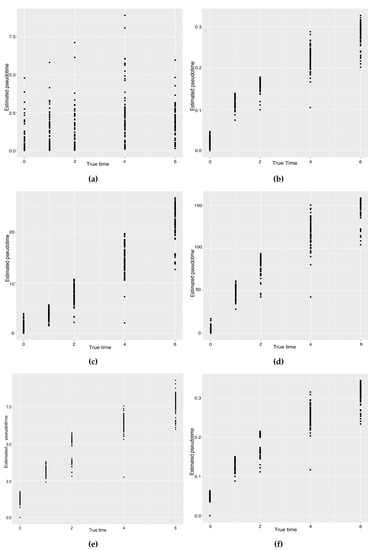
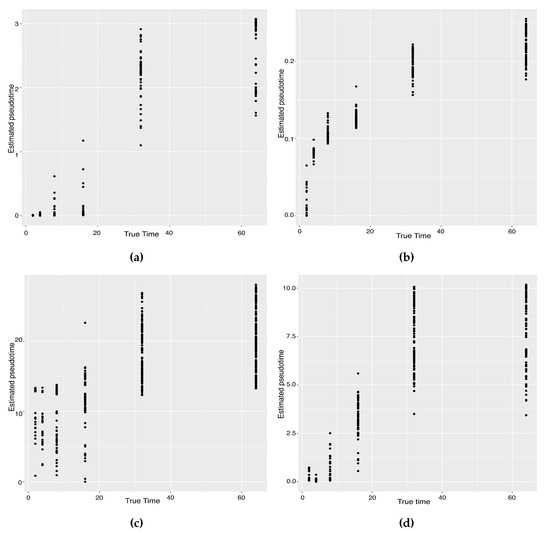
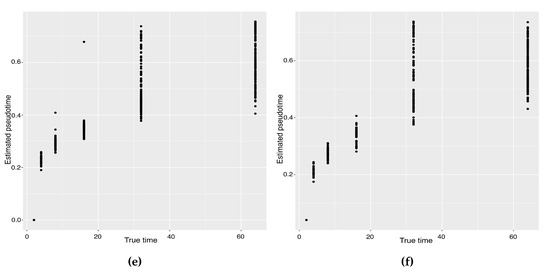
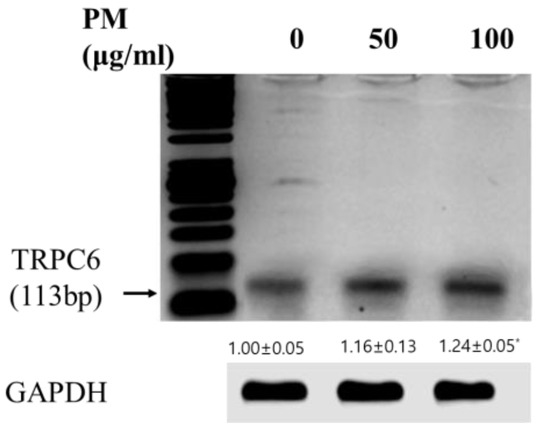
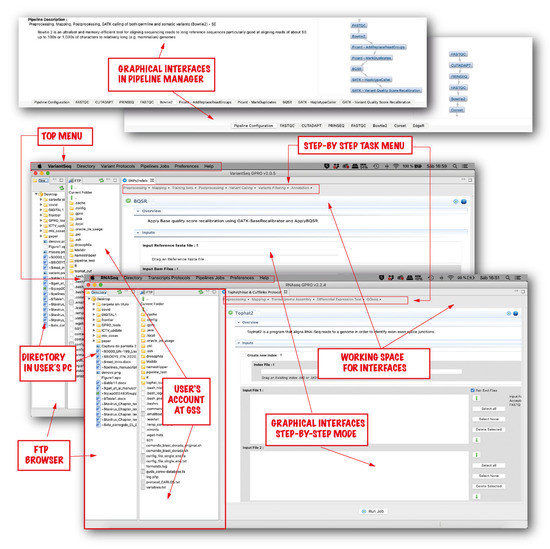
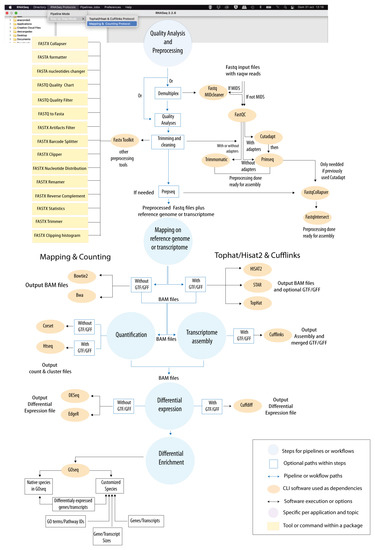
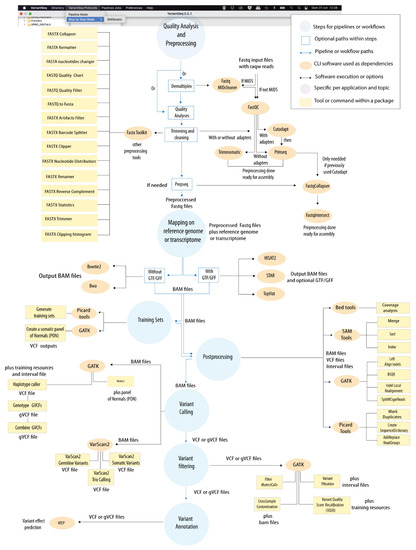
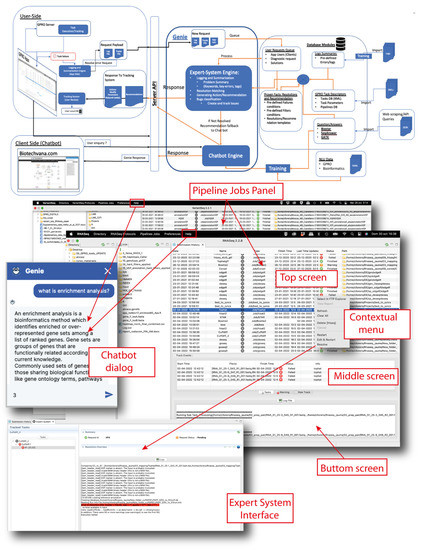
Background: Alfalfa, the most economically important forage legume worldwide, features modest genetic progress due to long selection cycles and the extent of the non-additive genetic variance associated with its autotetraploid genome. Methods: To improve the efficiency of genomic selection in alfalfa, we explored the effects of genome parametrization (as tetraploid and diploid dosages, plus allele ratios) and SNP marker subsetting (all available SNPs, only genic regions, and only non-genic regions) on genomic regressions, together with various levels of filtering on reading depth and missing rates. We used genotyping by sequencing-generated data and focused on traits of different genetic complexity, i.e., dry biomass yield in moisture-favorable (FE) and drought stress (SE) environments, leaf size, and the onset of flowering, which were assessed in 143 genotyped plants from a genetically broad European reference population and their phenotyped half-sib progenies. Results: On average, the allele ratio improved the predictive ability compared with other genome parametrizations (+7.9% vs. tetraploid dosage, +12.6% vs. diploid dosage), while using all the SNPs offered an advantage compared with any specific SNP subsetting (+3.7% vs. genic regions, +7.6% vs. non-genic regions). However, when focusing on specific traits, different combinations of genome parametrization and subsetting achieved better performances. We also released Legpipe2, an SNP calling pipeline tailored for reduced representation (GBS, RAD) in medium-sized genotyping experiments.
Full article

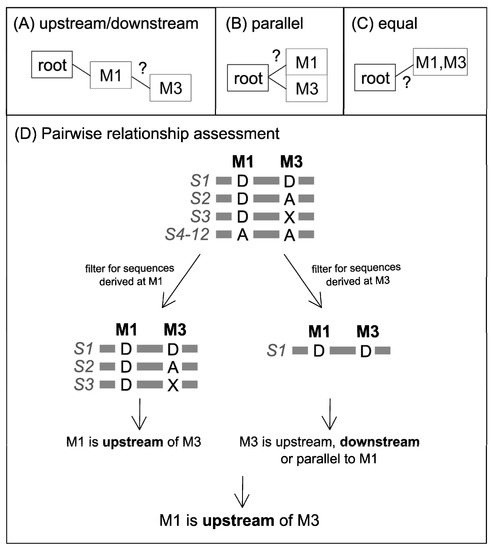
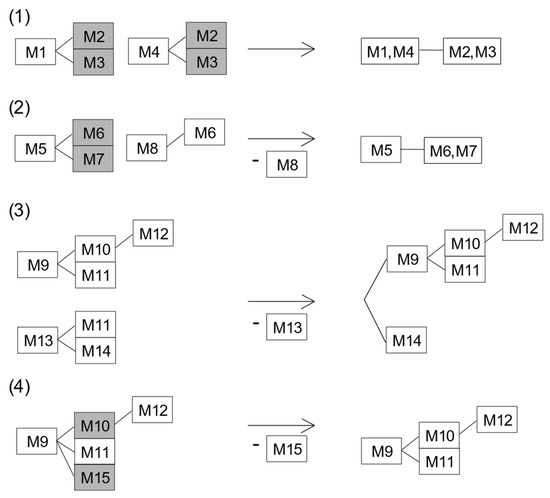
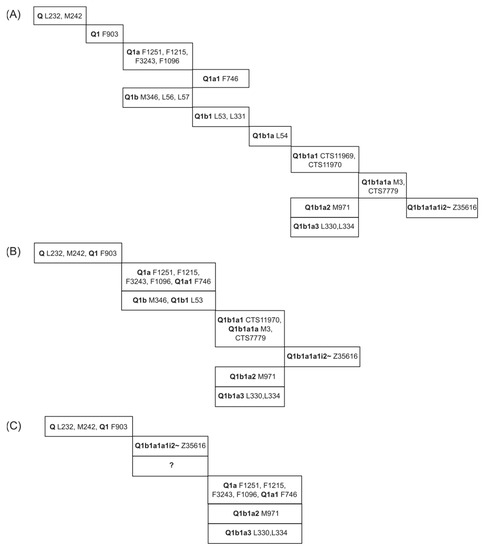
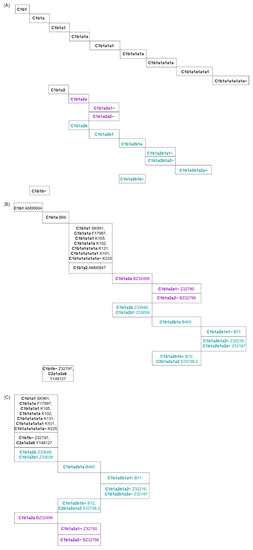
Figure 1
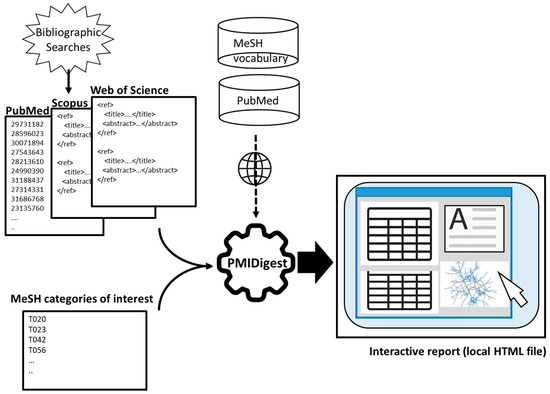

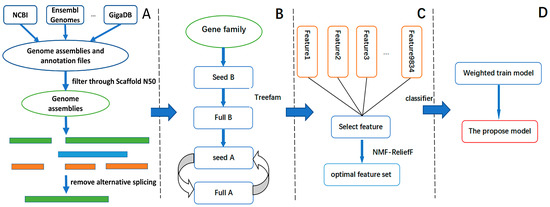

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}