
Detecting Melanocortin 1 Receptor Gene’s SNPs by CRISPR/enAsCas12a


Abstract
:1. Introduction
2. Materials and Methods
2.1. The Expression and Purification of enAsCas12a and LbCas12a
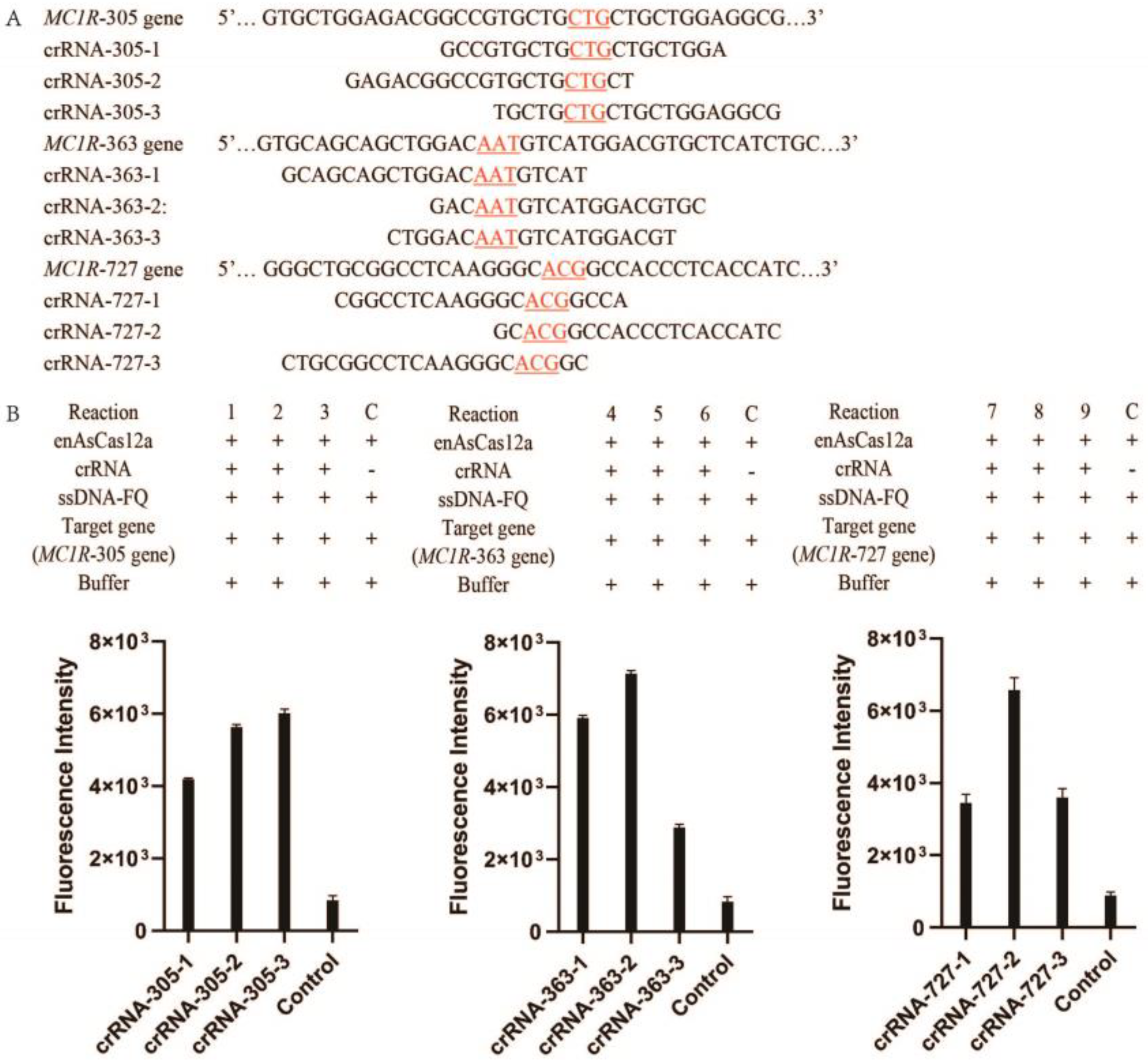
2.2. The Selection of Target Genes of MC1R
2.3. The RNA Transcription In Vitro
2.4. Optimization of the CRISPR/enAsCas12a Detection Assay
2.5. The CRISPR/enAsCas12a Detect the MC1R Gene and the MC1R Gene with SNP
2.6. Statistical Analysis
3. Results
3.1. The Detection of the MC1R Gene by CRISPR/enAsCas12a System
3.2. Optimization of the CRISPR/enAsCas12a Assay System
3.3. The SNPs of MC1R Detection by CRISPR/enAsCas12a
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Aman, R.; Mahas, A.; Mahfouz, M. Nucleic Acid Detection Using CRISPR/Cas Biosensing Technologies. ACS Synth. Biol. 2020, 9, 1226–1233. [Google Scholar] [CrossRef]
- Chen, J.S.; Ma, E.; Harrington, L.B.; Da Costa, M.; Tian, X.; Palefsky, J.M.; Doudna, J.A. CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science 2018, 360, 436–439. [Google Scholar] [CrossRef]
- Xie, S.; Tao, D.; Fu, Y.; Xu, B.; Tang, Y.; Steinaa, L.; Hemmink, J.D.; Pan, W.; Huang, X.; Nie, X.; et al. Rapid Visual CRISPR Assay: A Naked-Eye Colorimetric Detection Method for Nucleic Acids Based on CRISPR/Cas12a and a Convolutional Neural Network. ACS Synth. Biol. 2022, 11, 383–396. [Google Scholar] [CrossRef]
- Fozouni, P.; Son, S.; Diaz de Leon Derby, M.; Knott, G.J.; Gray, C.N.; D’Ambrosio, M.V.; Zhao, C.; Switz, N.A.; Kumar, G.R.; Stephens, S.I.; et al. Amplification-free detection of SARS-CoV-2 with CRISPR-Cas13a and mobile phone microscopy. Cell 2021, 184, 323–333.e329. [Google Scholar] [CrossRef]
- Soh, J.H.; Balleza, E.; Abdul Rahim, M.N.; Chan, H.M.; Mohd Ali, S.; Chuah, J.K.C.; Edris, S.; Atef, A.; Bahieldin, A.; Ying, J.Y.; et al. CRISPR-based systems for sensitive and rapid on-site COVID-19 diagnostics. Trends Biotechnol. 2022, 40, 1346–1360. [Google Scholar] [CrossRef]
- Wang, J.; Xia, Q.; Wu, J.; Lin, Y.; Ju, H. A sensitive electrochemical method for rapid detection of dengue virus by CRISPR/Cas13a-assisted catalytic hairpin assembly. Anal. Chim. Acta 2021, 1187, 339131. [Google Scholar] [CrossRef]
- Xu, B.; Gong, P.; Zhang, Y.; Wang, Y.; Tao, D.; Fu, L.; Khazalwa, E.M.; Liu, H.; Zhao, S.; Zhang, X.; et al. A one-tube rapid visual CRISPR assay for the field detection of Japanese encephalitis virus. Virus Res. 2022, 319, 198869. [Google Scholar] [CrossRef]
- Liu, S.; Tao, D.; Liao, Y.; Yang, Y.; Sun, S.; Zhao, Y.; Yang, P.; Tang, Y.; Chen, B.; Liu, Y.; et al. Highly Sensitive CRISPR/Cas12a-Based Fluorescence Detection of Porcine Reproductive and Respiratory Syndrome Virus. ACS Synth. Biol. 2021, 10, 2499–2507. [Google Scholar] [CrossRef]
- Ghosh, N.; Saha, I.; Sharma, N. Palindromic target site identification in SARS-CoV-2, MERS-CoV and SARS-CoV-1 by adopting CRISPR-Cas technique. Gene 2022, 818, 146136. [Google Scholar] [CrossRef]
- Talwar, C.S.; Park, K.H.; Ahn, W.C.; Kim, Y.S.; Kwon, O.S.; Yong, D.; Kang, T.; Woo, E. Detection of Infectious Viruses Using CRISPR-Cas12-Based Assay. Biosensors 2021, 11, 301. [Google Scholar] [CrossRef]
- Ma, P.; Meng, Q.; Sun, B.; Zhao, B.; Dang, L.; Zhong, M.; Liu, S.; Xu, H.; Mei, H.; Liu, J.; et al. MeCas12a, a Highly Sensitive and Specific System for COVID-19 Detection. Adv. Sci. 2020, 7, 2001300. [Google Scholar] [CrossRef] [PubMed]
- Swarts, D.C.; Jinek, M. Mechanistic Insights into the cis- and trans-Acting DNase Activities of Cas12a. Mol. Cell 2019, 73, 589–600.e584. [Google Scholar] [CrossRef] [PubMed]
- Ooi, K.H.; Liu, M.M.; Tay, J.W.D.; Teo, S.Y.; Kaewsapsak, P.; Jin, S.; Lee, C.K.; Hou, J.; Maurer-Stroh, S.; Lin, W.; et al. An engineered CRISPR-Cas12a variant and DNA-RNA hybrid guides enable robust and rapid COVID-19 testing. Nat. Commun. 2021, 12, 1739. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Smith, B.M.; Jain, P.K. Enhancement of trans-cleavage activity of Cas12a with engineered crRNA enables amplified nucleic acid detection. Nat. Commun. 2020, 11, 4906. [Google Scholar] [CrossRef] [PubMed]
- Huyke, D.A.; Ramachandran, A.; Bashkirov, V.I.; Kotseroglou, E.K.; Kotseroglou, T.; Santiago, J.G. Enzyme Kinetics and Detector Sensitivity Determine Limits of Detection of Amplification-Free CRISPR-Cas12 and CRISPR-Cas13 Diagnostics. Anal. Chem. 2022, 94, 9826–9834. [Google Scholar] [CrossRef]
- Ramachandran, A.; Santiago, J.G. CRISPR Enzyme Kinetics for Molecular Diagnostics. Anal. Chem. 2021, 93, 7456–7464. [Google Scholar] [CrossRef]
- Rossetti, M.; Merlo, R.; Bagheri, N.; Moscone, D.; Valenti, A.; Saha, A.; Arantes, P.R.; Ippodrino, R.; Ricci, F.; Treglia, I.; et al. Enhancement of CRISPR/Cas12a trans-cleavage activity using hairpin DNA reporters. Nucleic Acids Res. 2022, 50, 8377–8391. [Google Scholar] [CrossRef]
- Zhang, L.; Jiang, Z.; Zhou, Z.; Sun, J.; Yan, S.; Gao, W.; Shao, Y.; Bai, Y.; Wu, Y.; Yan, Z.; et al. A TaqMan Probe-Based Multiplex Real-Time PCR for Simultaneous Detection of Porcine Epidemic Diarrhea Virus Subtypes G1 and G2, and Porcine Rotavirus Groups A and C. Viruses 2022, 14, 1819. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.T.; Alam, A.R.U.; Sakib, N.; Hasan, M.S.; Chakrovarty, T.; Tawyabur, M.; Islam, O.K.; Al-Emran, H.M.; Jahid, M.I.K.; Anwar Hossain, M. A rapid and cost-effective multiplex ARMS-PCR method for the simultaneous genotyping of the circulating SARS-CoV-2 phylogenetic clades. J. Med. Virol. 2021, 93, 2962–2970. [Google Scholar] [CrossRef]
- Dema, A.; Ganji, V.K.; Yella, N.R.; Putty, K. A novel one-step amplification refractory mutation system PCR (ARMS-PCR) for differentiation of canine parvovirus-2 variants. Virus Genes 2021, 57, 426–433. [Google Scholar] [CrossRef]
- Alyethodi, R.R.; Singh, U.; Kumar, S.; Alex, R.; Sengar, G.S.; Raja, T.V.; Deb, R.; Prakash, B. Designing, optimization, and validation of whole blood direct T-ARMS PCR for precise and rapid genotyping of complex vertebral malformation in cattle. BMC Biotechnol. 2021, 21, 36. [Google Scholar] [CrossRef]
- Bookari, K.; Arrish, J.; Zaher, S.; Alkhalaf, M.; Alharbi, M.; Alkhattaf, N.; Harb, Z.; Al Hinai, E.; Alanqodi, N.; Almajed, S.; et al. A Snapshot of the Experience of Dietitians during the COVID-19 Crisis in Five Arab Countries: Findings from a Regional Cross-Sectional Study. Nutrients 2022, 14, 4904. [Google Scholar] [CrossRef]
- Li, L.; Li, S.; Wu, N.; Wu, J.; Wang, G.; Zhao, G.; Wang, J. HOLMESv2: A CRISPR-Cas12b-Assisted Platform for Nucleic Acid Detection and DNA Methylation Quantitation. ACS Synth. Biol. 2019, 8, 2228–2237. [Google Scholar] [CrossRef]
- Chen, Y.; Mei, Y.; Jiang, X. Universal and high-fidelity DNA single nucleotide polymorphism detection based on a CRISPR/Cas12a biochip. Chem. Sci. 2021, 12, 4455–4462. [Google Scholar] [CrossRef]
- Wang, M.; Liu, X.; Yang, J.; Wang, Z.; Wang, H.; Wang, X. CRISPR/Cas12a-based biosensing platform for the on-site detection of single-base mutants in gene-edited rice. Front. Plant Sci. 2022, 13, 944295. [Google Scholar] [CrossRef]
- Kijas, J.M.; Wales, R.; Tornsten, A.; Chardon, P.; Moller, M.; Andersson, L. Melanocortin receptor 1 (MC1R) mutations and coat color in pigs. Genetics 1998, 150, 1177–1185. [Google Scholar] [CrossRef]
- Ji, R.L.; Tao, Y.X. Melanocortin-1 receptor mutations and pigmentation: Insights from large animals. Prog. Mol. Biol. Transl. Sci. 2022, 189, 179–213. [Google Scholar] [CrossRef]
- Zhong, H.; Zhang, J.; Tan, C.; Shi, J.; Yang, J.; Cai, G.; Wu, Z.; Yang, H. Pig Coat Color Manipulation by MC1R Gene Editing. Int. J. Mol. Sci. 2022, 23, 356. [Google Scholar] [CrossRef]
- Wang, W.; Guo, D.Y.; Lin, Y.J.; Tao, Y.X. Melanocortin Regulation of Inflammation. Front. Endocrinol. 2019, 10, 683. [Google Scholar] [CrossRef]
- Switonski, M.; Mankowska, M.; Salamon, S. Family of melanocortin receptor (MCR) genes in mammals-mutations, polymorphisms and phenotypic effects. J. Appl. Genet. 2013, 54, 461–472. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Zhu, B.; Yin, C.; Liu, W.; Han, C.; Chen, B.; Liu, T.; Li, X.; Chen, X.; Li, C.; et al. Palmitoylation-dependent activation of MC1R prevents melanomagenesis. Nature 2017, 549, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Almathen, F.; Elbir, H.; Bahbahani, H.; Mwacharo, J.; Hanotte, O. Polymorphisms in MC1R and ASIP Genes are Associated with Coat Color Variation in the Arabian Camel. J. Hered. 2018, 109, 700–706. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Allete | Coat Color | Codon | ||
|---|---|---|---|---|
| 102 | 121 | 243 | ||
| MC1R*1(E+) | Wild type, European wild boar | CTG Leu | AAT Asn | GCG Ala |
| MC1R*1(E+) | Wild type, Japanese wild boar | --- - | --C - | --- - |
| MC1R*2(ED1) | Dominant black | -C- - | --C - | --A - |
| MC1R*4(e) | Recessive red | --- - | --- - | A-- Thr |
| Gene | Sequence |
|---|---|
| MC1R-305 gene | …5’-GAGACGGCCGTGCTGCTGCTGCTGGAGGCGGGC-3’… |
| MC1R-T305C SNP | …5’-GAGACGGCCGTGCTGCcGCTGCTGGAGGCGGGC-3’… |
| MC1R-363 gene | …5’-GTGCAGCAGCTGGACAATGTCATGGACGTGCTC-3’… |
| MC1R-T363C SNP | …5’-GTGCAGCAGCTGGACAAcGTCATGGACGTGCTC-3’… |
| MC1R-727 gene | …5’-TGCGGCCTCAAGGGCACGGCCACCCTCACCATC-3’… |
| MC1R-A727G SNP | …5’-TGCGGCCTCAAGGGCgCGGCCACCCTCACCATC -3’… |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, W.; Tao, D.; Xu, B.; Zheng, Y.; Zhao, S. Detecting Melanocortin 1 Receptor Gene’s SNPs by CRISPR/enAsCas12a. Genes 2023, 14, 394. https://doi.org/10.3390/genes14020394
Yang W, Tao D, Xu B, Zheng Y, Zhao S. Detecting Melanocortin 1 Receptor Gene’s SNPs by CRISPR/enAsCas12a. Genes. 2023; 14(2):394. https://doi.org/10.3390/genes14020394
Chicago/Turabian StyleYang, Wei, Dagang Tao, Bingrong Xu, Yueting Zheng, and Shuhong Zhao. 2023. "Detecting Melanocortin 1 Receptor Gene’s SNPs by CRISPR/enAsCas12a" Genes 14, no. 2: 394. https://doi.org/10.3390/genes14020394