
A Role for the Interactions between Polδ and PCNA Revealed by Analysis of pol3-01 Yeast Mutants

Abstract
:1. Introduction
2. Materials and Methods
2.1. Yeast Strains
2.2. Plasmids
2.3. Media and Growth Conditions
2.4. Yeast Two-Hybrid (Y2H) Assay
2.5. Fluctuation Test
2.6. Fractionation Analysis
2.7. Protein Extraction and Immunoprecipitation Assays
3. Results
3.1. Increased Mutagenesis and Unequal Sister Chromatid Recombination in Pol3-01 Mutants
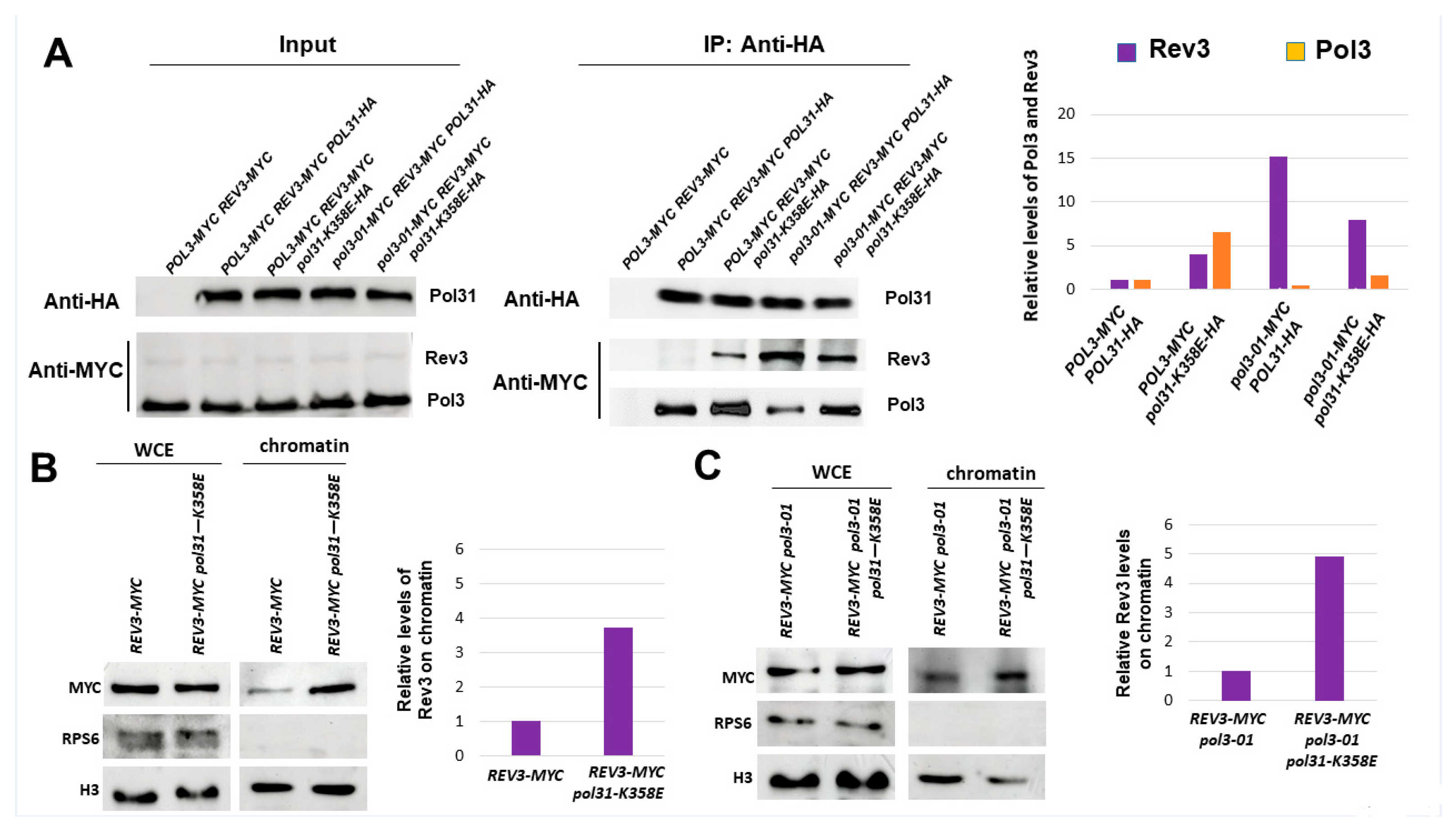
3.2. Reduced Interaction between Pol3-01 and PCNA
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pavlov, Y.I.; Frahm, C.; Nick McElhinny, S.A.; Niimi, A.; Suzuki, M.; Kunkel, T.A. Evidence that errors made by DNA polymerase α are corrected by DNA polymerase δ. Curr. Biol. 2006, 16, 202–207. [Google Scholar] [CrossRef] [PubMed]
- Burgers, P.M.J.; Kunkel, T.A. Eukaryotic DNA Replication Fork. Annu. Rev. Biochem. 2017, 86, 417–438. [Google Scholar] [CrossRef] [PubMed]
- Pursell, Z.F.; Isoz, I.; Lundstrom, E.B.; Johansson, E.; Kunkel, T.A. Yeast DNA polymerase epsilon participates in leading-strand DNA replication. Science 2007, 317, 127–130. [Google Scholar] [CrossRef] [PubMed]
- Nick McElhinny, S.A.; Gordenin, D.A.; Stith, C.M.; Burgers, P.M.; Kunkel, T.A. Division of labor at the eukaryotic replication fork. Mol. Cell 2008, 30, 137–144. [Google Scholar] [CrossRef]
- Bauer, G.A.; Burgers, P.M. The yeast analog of mammalian cyclin/proliferating-cell nuclear antigen interacts with mammalian DNA polymerase delta. Proc. Natl. Acad. Sci. USA 1988, 85, 7506–7510. [Google Scholar] [CrossRef]
- Jonsson, Z.O.; Hindges, R.; Hubscher, U. Regulation of DNA replication and repair proteins through interaction with the front side of proliferating cell nuclear antigen. EMBO J. 1998, 17, 2412–2425. [Google Scholar] [CrossRef]
- Warbrick, E. The puzzle of PCNA’s many partners. BioEssays 2000, 22, 997–1006. [Google Scholar] [CrossRef]
- Boulet, A.; Simon, M.; Faye, G.; Bauer, G.A.; Burgers, P.M. Structure and function of the Saccharomyces cerevisiae CDC2 gene encoding the large subunit of DNA polymerase III. EMBO J 1989, 8, 1849–1854. [Google Scholar] [CrossRef]
- Gerik, K.J.; Li, X.; Pautz, A.; Burgers, P.M. Characterization of the two small subunits of Saccharomyces cerevisiae DNA polymerase δ. J. Biol. Chem. 1998, 273, 19747–19755. [Google Scholar] [CrossRef]
- Johansson, E.; Majka, J.; Burgers, P.M. Structure of DNA polymerase δ from Saccharomyces cerevisiae. J. Biol. Chem. 2001, 276, 43824–43828. [Google Scholar] [CrossRef] [Green Version]
- Huang, M.E.; Cadieu, E.; Souciet, J.L.; Galibert, F. Disruption of six novel yeast genes reveals three genes essential for vegetative growth and one required for growth at low temperature. Yeast 1997, 13, 1181–1194. [Google Scholar] [CrossRef]
- Johansson, E.; Garg, P.; Burgers, P.M. The Pol32 subunit of DNA polymerase δ contains separable domains for processive replication and proliferating cell nuclear antigen (PCNA) binding. J. Biol. Chem. 2004, 279, 1907–1915. [Google Scholar] [CrossRef]
- Jain, R.; Rice, W.J.; Malik, R.; Johnson, R.E.; Prakash, L.; Prakash, S.; Ubarretxena-Belandia, I.; Aggarwal, A.K. Cryo-EM structure and dynamics of eukaryotic DNA polymerase δ holoenzyme. Nat. Struct. Mol. Biol. 2019, 26, 955–962. [Google Scholar] [CrossRef]
- Acharya, N.; Klassen, R.; Johnson, R.E.; Prakash, L.; Prakash, S. PCNA binding domains in all three subunits of yeast DNA polymerase δ modulate its function in DNA replication. Proc. Natl. Acad. Sci. USA 2011, 108, 17927–17932. [Google Scholar] [CrossRef]
- Burkovics, P.; Sebesta, M.; Sisakova, A.; Plault, N.; Szukacsov, V.; Robert, T.; Pinter, L.; Marini, V.; Kolesar, P.; Haracska, L.; et al. Srs2 mediates PCNA-SUMO-dependent inhibition of DNA repair synthesis. EMBO J. 2013, 32, 742–755. [Google Scholar] [CrossRef]
- Arbel, M.; Liefshitz, B.; Kupiec, M. How yeast cells deal with stalled replication forks. Curr. Genet. 2020, 66, 911–915. [Google Scholar] [CrossRef]
- Arbel, M.; Liefshitz, B.; Kupiec, M. DNA damage bypass pathways and their effect on mutagenesis in yeast. FEMS Microbiol. Rev. 2021, 45, fuaa038. [Google Scholar] [CrossRef]
- Bailly, V.; Lauder, S.; Prakash, S.; Prakash, L. Yeast DNA repair proteins Rad6 and Rad18 form a heterodimer that has ubiquitin conjugating, DNA binding, and ATP hydrolytic activities. J. Biol. Chem. 1997, 272, 23360–23365. [Google Scholar] [CrossRef]
- Hoege, C.; Pfander, B.; Moldovan, G.L.; Pyrowolakis, G.; Jentsch, S. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature 2002, 419, 135–141. [Google Scholar] [CrossRef]
- Nelson, J.R.; Lawrence, C.W.; Hinkle, D.C. Thymine-thymine dimer bypass by yeast DNA polymerase ζ. Science 1996, 272, 1646–1649. [Google Scholar] [CrossRef]
- Cassier, C.; Chanet, R.; Henriques, J.A.; Moustacchi, E. The effects of three PSO genes on induced mutagenesis: A novel class of mutationally defective yeast. Genetics 1980, 96, 841–857. [Google Scholar] [CrossRef] [PubMed]
- Quah, S.K.; von Borstel, R.C.; Hastings, P.J. The origin of spontaneous mutation in Saccharomyces cerevisiae. Genetics 1980, 96, 819–839. [Google Scholar] [CrossRef] [PubMed]
- Gan, G.N.; Wittschieben, J.P.; Wittschieben, B.O.; Wood, R.D. DNA polymerase zeta (pol ζ) in higher eukaryotes. Cell Res. 2008, 18, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Goodman, M.F. Error-prone repair DNA polymerases in prokaryotes and eukaryotes. Annu. Rev. Biochem. 2002, 71, 17–50. [Google Scholar] [CrossRef]
- Branzei, D.; Szakal, B. DNA damage tolerance by recombination: Molecular pathways and DNA structures. DNA Repair 2016, 44, 68–75. [Google Scholar] [CrossRef]
- Zhang, H.; Lawrence, C.W. The error-free component of the RAD6/RAD18 DNA damage tolerance pathway of budding yeast employs sister-strand recombination. Proc. Natl. Acad. Sci. USA 2005, 102, 15954–15959. [Google Scholar] [CrossRef]
- Ulrich, H.D.; Jentsch, S. Two RING finger proteins mediate cooperation between ubiquitin-conjugating enzymes in DNA repair. EMBO J. 2000, 19, 3388–3397. [Google Scholar] [CrossRef]
- Torres-Ramos, C.A.; Prakash, S.; Prakash, L. Requirement of RAD5 and MMS2 for postreplication repair of UV-damaged DNA in Saccharomyces cerevisiae. Mol. Cell. Biol. 2002, 22, 2419–2426. [Google Scholar] [CrossRef]
- Church, D.N.; Briggs, S.E.; Palles, C.; Domingo, E.; Kearsey, S.J.; Grimes, J.M.; Gorman, M.; Martin, L.; Howarth, K.M.; Hodgson, S.V.; et al. DNA polymerase ε and δ exonuclease domain mutations in endometrial cancer. Hum. Mol. Genet. 2013, 22, 2820–2828. [Google Scholar] [CrossRef]
- Palles, C.; Cazier, J.B.; Howarth, K.M.; Domingo, E.; Jones, A.M.; Broderick, P.; Kemp, Z.; Spain, S.L.; Guarino, E.; Salguero, I.; et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat. Genet. 2013, 45, 136–144. [Google Scholar] [CrossRef] [Green Version]
- Morrison, A.; Johnson, A.L.; Johnston, L.H.; Sugino, A. Pathway correcting DNA replication errors in Saccharomyces cerevisiae. EMBO J. 1993, 12, 1467–1473. [Google Scholar] [CrossRef]
- Kadyk, L.C.; Hartwell, L.H. Sister chromatids are preferred over homologs as substrates for recombinational repair in Saccharomyces cerevisiae. Genetics 1992, 132, 387–402. [Google Scholar] [CrossRef]
- Lea, D.E.; Coulson, C.A. The distribution of the numbers of mutants in bacterial populations. J. Genet. 1949, 49, 264–285. [Google Scholar] [CrossRef]
- Simon, M.; Giot, L.; Faye, G. The 3′ to 5′ exonuclease activity located in the DNA polymerase delta subunit of Saccharomyces cerevisiae is required for accurate replication. EMBO J. 1991, 10, 2165–2170. [Google Scholar] [CrossRef]
- Jin, Y.H.; Obert, R.; Burgers, P.M.; Kunkel, T.A.; Resnick, M.A.; Gordenin, D.A. The 3′-->5′ exonuclease of DNA polymerase δ can substitute for the 5′ flap endonuclease Rad27/Fen1 in processing Okazaki fragments and preventing genome instability. Proc. Natl. Acad. Sci. USA 2001, 98, 5122–5127. [Google Scholar] [CrossRef]
- Tran, H.T.; Gordenin, D.A.; Resnick, M.A. The 3′-->5′ exonucleases of DNA polymerases delta and epsilon and the 5'-->3' exonuclease Exo1 have major roles in postreplication mutation avoidance in Saccharomyces cerevisiae. Mol. Cell. Biol. 1999, 19, 2000–2007. [Google Scholar] [CrossRef]
- Lang, G.I.; Murray, A.W. Estimating the per-base-pair mutation rate in the yeast Saccharomyces cerevisiae. Genetics 2008, 178, 67–82. [Google Scholar] [CrossRef]
- Tran, H.T.; Keen, J.D.; Kricker, M.; Resnick, M.A.; Gordenin, D.A. Hypermutability of homonucleotide runs in mismatch repair and DNA polymerase proofreading yeast mutants. Mol. Cell. Biol. 1997, 17, 2859–2865. [Google Scholar] [CrossRef]
- Johnson, R.E.; Prakash, L.; Prakash, S. Pol31 and Pol32 subunits of yeast DNA polymerase delta are also essential subunits of DNA polymerase zeta. Proc. Natl. Acad. Sci. USA 2012, 109, 12455–12460. [Google Scholar] [CrossRef]
- Giot, L.; Chanet, R.; Simon, M.; Facca, C.; Faye, G. Involvement of the yeast DNA polymerase delta in DNA repair in vivo. Genetics 1997, 146, 1239–1251. [Google Scholar] [CrossRef]
- Giot, L.; Simon, M.; Dubois, C.; Faye, G. Suppressors of thermosensitive mutations in the DNA polymerase delta gene of Saccharomyces cerevisiae. Mol. Gen. Genet. 1995, 246, 212–222. [Google Scholar] [CrossRef] [PubMed]
- Volkova, N.V.; Meier, B.; Gonzalez-Huici, V.; Bertolini, S.; Gonzalez, S.; Vohringer, H.; Abascal, F.; Martincorena, I.; Campbell, P.J.; Gartner, A.; et al. Mutational signatures are jointly shaped by DNA damage and repair. Nat. Commun. 2020, 11, 2169. [Google Scholar] [CrossRef] [PubMed]
- Brutlag, D.; Kornberg, A. Enzymatic synthesis of deoxyribonucleic acid. 36. A proofreading function for the 3′ leads to 5′ exonuclease activity in deoxyribonucleic acid polymerases. J. Biol. Chem. 1972, 247, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Kunkel, T.A. Exonucleolytic proofreading. Cell 1988, 53, 837–840. [Google Scholar] [CrossRef]
- Jin, Y.H.; Garg, P.; Stith, C.M.; Al-Refai, H.; Sterling, J.F.; Murray, L.J.; Kunkel, T.A.; Resnick, M.A.; Burgers, P.M.; Gordenin, D.A. The multiple biological roles of the 3′-->5′ exonuclease of Saccharomyces cerevisiae DNA polymerase delta require switching between the polymerase and exonuclease domains. Mol. Cell. Biol. 2005, 25, 461–471. [Google Scholar] [CrossRef]
- Morrison, A.; Sugino, A. The 3′-->5′ exonucleases of both DNA polymerases delta and epsilon participate in correcting errors of DNA replication in Saccharomyces cerevisiae. Mol. Gen. Genet. 1994, 242, 289–296. [Google Scholar] [CrossRef]
- Hendel, A.; Krijger, P.H.; Diamant, N.; Goren, Z.; Langerak, P.; Kim, J.; Reissner, T.; Lee, K.Y.; Geacintov, N.E.; Carell, T.; et al. PCNA ubiquitination is important, but not essential for translesion DNA synthesis in mammalian cells. PLoS Genet. 2011, 7, e1002262. [Google Scholar] [CrossRef]
- Tellier-Lebegue, C.; Dizet, E.; Ma, E.; Veaute, X.; Coic, E.; Charbonnier, J.B.; Maloisel, L. The translesion DNA polymerases Pol ζ and Rev1 are activated independently of PCNA ubiquitination upon UV radiation in mutants of DNA polymerase δ. PLoS Genet. 2017, 13, e1007119. [Google Scholar] [CrossRef]
- Sakofsky, C.J.; Ayyar, S.; Deem, A.K.; Chung, W.H.; Ira, G.; Malkova, A. Translesion Polymerases Drive Microhomology-Mediated Break-Induced Replication Leading to Complex Chromosomal Rearrangements. Mol. Cell 2015, 60, 860–872. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number | Name | Genotype |
|---|---|---|
| 18071 | E134 | MAT@ ade5-1,lys2::InsEa14,trp1-289,his7-2,leu2-3,112,ura3-52 |
| 19779 | E134 POL3-MYC POL30-FLAG | MATa ade5-1,lys2::InsEa14,trp1-289,his7-2,leu2-3,112,ura3-52, POL3-MYC-TRP1 POL30-FLAG-KanMX |
| 19780 | E134 POL30-FLAG POL3-01-MYC | MATa ade5-1,lys2::InsEa14,trp1-289,his7-2,leu2-3,112,ura3-52, pol3-01-MYC-TRP1 POL30-FLAG-KanMX |
| BLX1 | E134 rev3 | MAT@ ade5-1,lys2::InsEa14,trp1-289,his7-2,leu2-3,112,ura3-52 rev3:KanMX del |
| BLX2 | E134 rev3 pol3-01-V5 | MAT@ ade5-1,lys2::InsEa14,trp1-289,his7-2,leu2-3,112,ura3-52 pol3-01-V5-KanMX rev3:KanMX |
| BLX3 | E134 POL3-V5 | MAT@ ade5-1,lys2::InsEa14,trp1-289,his7-2,leu2-3,112,ura3-52 POL3-V5-KanMX |
| BLX4 | E134 pol3-01-V5 | MAT@ ade5-1,lys2::InsEa14,trp1-289,his7-2,leu2-3,112,ura3-52 pol3-01- V5-KanMX |
| BLX5 | E134 pol3-01-pip32-V5 | MAT@ ade5-1,lys2::InsEa14,trp1-289,his7-2,leu2-3,112,ura3-52 pol3-01-pip32-V5;KanMX rev3:KanMX |
| BLX6 | E134 POL3-MYC REV3-MYC pol31-HA | MAT@ ade5-1,lys2::InsEa14,trp1-289,his7-2,leu2-3,112,ura3-52 POL3-MYC-TRP1 REV3-MYC-HygMX pol31-HA-NatMX |
| BLX7 | E134 pol3-01-MYC REV3-MYC pol31-HA | MAT@ ade5-1,lys2::InsEa14,trp1-289,his7-2,leu2-3,112,ura3-52 pol3-01-MYC-TRP1 REV3-MYC-HygMX pol31-HA-NatMX |
| BLX8 | E134 POL3-MYC REV3-MYC pol31-K358E-HA | MAT@ ade5-1,lys2::InsEa14,trp1-289,his7-2,leu2-3,112,ura3-52 POL3-MYC-TRP1 REV3-MYC-HygMX pol31-K358E-HA-NatMX |
| BLX9 | E134 pol3-01-MYC REV3-MYC pol31-K358E-HA | MAT@ ade5-1,lys2::InsEa14,trp1-289,his7-2,leu2-3,112,ura3-52 pol3-01-MYC-TRP1 REV3-MYC-HygMX pol31-K358E-HA-NatMX |
| BLX10 | E134 POL3- MYC POL30-FLAG | MAT@ ade5-1,lys2::InsEa14,trp1-289,his7-2,leu2-3,112,ura3-52 POL3- MYC-TRP1 POL30-FLAG-KanMX |
| BLX11 | E134 POL3-pip32-MYC POL30-FLAG | MAT@ ade5-1,lys2::InsEa14,trp1-289,his7-2,leu2-3,112,ura3-52 POL3-pip32-MYC-TRP1 POL30-FLAG-KanMX |
| BLX12 | E134 pol3-01- MYC POL30-FLAG | MAT@ ade5-1,lys2::InsEa14,trp1-289,his7-2,leu2-3,112,ura3-52 pol3-01-MYC-TRP1 POL30-FLAG-KanMX |
| BLX13 | E134 pol3-01-pip32-MYC POL30-FLAG | MAT@ ade5-1,lys2::InsEa14,trp1-289,his7-2,leu2-3,112,ura3-52 pol3-01-pip32-MYC-TRP1 POL30-FLAG-KanMX |
| 18178 | PJ694 | MATa MAT trp1-901 leu2-3,112 ura3-52 his3-200 gal4del gal80del GAL2-ADE2 LYS2:: GAL1-HIS3 met2::GAL7-lacZ |
| 19850 | BLS2 | MATa ura3-52, trp1del1, leu2-3, 112 lys2::LTR, His3::lys::ura3-, can1-101, ade2-o ade3,ade3::ura3-::ade3 in chrom. III |
| 18740 | BLS2 pol3-01 | MATa ura3-52, trp1del1, leu2-3, lys2::ty::sup, His3::lys::ura3, can101, ade2-o, ade3 ade3::ura3-::ade3 in chrom. III pol3-01-myc-TRP1 |
| 19217 | BLS2 rev3:KanMX | MATa ura3-52, trp1del1, leu2-3, 112 lys2::LTR, His3::lys::ura3-, can1-101, ade2-o ade3,ade3::ura3-::ade3 in chrom. III rev3:KanMX |
| 19257 | BLS2 rev3 pol3-01 | MATa ura3-52, trp1del1, leu2-3, 112 lys2::LTR, His3::lys::ura3-, can1-101, ade2-o ade3,ade3::ura3-::ade3 in chrom. III rev3:KanMX pol3-01-myc-TRP1 |
| 19398 | BLS2 pol3-01-pip32 | MATa ura3-52, trp1del1, leu2-3, lys2::ty::sup, His3::lys::ura3, can101, ade2-o, ade3 ade3::ura3-::ade3 in chrom. III pol3-01-pip32-myc-TRP1 |
| 19781 | BLS2 REV3-Myc | MATa ura3-52, trp1del1, leu2-3, 112 lys2::LTR, His3::lys::ura3-, can1-101, ade2-o ade3, Ade3::ura3-::ade3 in chrom. III, REV3-MYC-HYGMX |
| 19782 | BLS2 pol3-01-Myc REV3-Myc | mk166- MATa ura3-52, trp1del1, leu2-3, 112 lys2::LTR, His3::lys::ura3-, can1-101, ade2-o ade3, Ade3::ura3-::ade3 in chrom. III, REV3-MYC-HYGMX, pol3-01-MYC-TRP1 |
| 20286 | BLS2 pol31-K358E | MAT@ ura3-52, trp1del1, leu2-3, 112 lys2::LTR, His3::lys::ura3-, can1-101, ade2-o ade3, Ade3::ura3-::ade3 in chrom. III, pol31-K358E |
| 20288 | BL2 REV3-MYC pol31-K358E | MAT@ ura3-52, trp1del1, leu2-3, 112 lys2::LTR, His3::lys::ura3-, can1-101, ade2-o ade3, plk19 Ade3::ura3-::ade3 in chrom. III, REV3-MYC-HYGMX, pol31-K358E |
| 20290 | BL2 REV3-MYC pol3-01 | MAT@ ura3-52, trp1del1, leu2-3, 112 lys2::LTR, His3::lys::ura3-, can1-101, ade2-o ade3, plk19 Ade3::ura3-::ade3 in chrom. III, REV3-MYC-HYGMX pol3-01-myc-TRP1 |
| 20292 | BL2 pol31-K358E pol3-01 | MAT@ ura3-52, trp1del1, leu2-3, 112 lys2::LTR, His3::lys::ura3-, can1-101, ade2-o ade3, Ade3::ura3-::ade3 in chrom. III, pol31-K358E pol3-01-myc-TRP1 |
| 20294 | BL2 REV3-MYC pol31-K358E pol3-01 | MAT@ ura3-52, trp1del1, leu2-3, 112 lys2::LTR, His3::lys::ura3-, can1-101, ade2-o ade3, plk19 Ade3::ura3-::ade3 in chrom. III, REV3-MYC-HYGMX pol3-01-myc-TRP1 pol31-K358E |
| 20338 | BLS2 pol3-pip32 #110 | MAT@ ura3-52, trp1del1, leu2-3, 112 lys2::LTR, His3::lys::ura3-, can1-101, ade2-o ade3, Ade3::ura3-::ade3 in chrom. III, pol3-pip32-MYC-TRP1 |
| 20354 | BLS2 POL3-MYC | MAT@ ura3-52, trp1del1, leu2-3, 112 lys2::LTR, His3::lys::ura3-, can1-101, ade2-o ade3, Ade3::ura3-::ade3 in chrom. III, POL3-MYC-TRP1 |
| 20355 | BLS2 POL3-MYC pol31-K358E | MAT@ ura3-52, trp1del1, leu2-3, 112 lys2::LTR, His3::lys::ura3-, can1-101, ade2-o ade3, Ade3::ura3-::ade3 in chrom. III, POL3-MYC-TRP1 pol31-K358E-NatMX |
| 20362 | BLS2 pol3-01-pip32 REV3-MYC | MAT@ ura3-52, trp1del1, leu2-3, 112 lys2::LTR, His3::lys::ura3-, can1-101, ade2-o ade3, plk19 Ade3::ura3-::ade3 in chrom. III, REV3-MYC-HYGMX, pol3-01-pip32-MYC-TRP1 |
| 4112 | pACT2 | Y2H Vector, with a LEU2 Yeast Marker, Containing the GAL4 AD (Amino Acids 768–881), and an HA Epitope tag |
| 1960 | opb63b | pACT2 carrying POL30 |
| 2349 | YIpAM26 | AmpR, pol3-01. Received from R. Kolodner as RDK3097, made by Sugino as YIpAM26, Morrison et al. EMBO, vol. 12 No. 4, 1993 |
| 1979 | pGBKT7 | Y2H vector, with a KanR bacterial selection and TRP1 yeast marker, containing an N-terminal Myc tag and the GAL4 DNA binding domain (BD) under ADH promoter |
| 4091 | PGBKT7 POL3 | POL3 cloned into pGBKT7 |
| 4093 | PGBKT7 pol3-pip32 | pol3-pip32 cloned into pGBKT7 |
| 4095 | PGBKT7 pol3-01 | pol3-01 cloned into pGBKT7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nir Heyman, S.; Golan, M.; Liefshitz, B.; Kupiec, M. A Role for the Interactions between Polδ and PCNA Revealed by Analysis of pol3-01 Yeast Mutants. Genes 2023, 14, 391. https://doi.org/10.3390/genes14020391
Nir Heyman S, Golan M, Liefshitz B, Kupiec M. A Role for the Interactions between Polδ and PCNA Revealed by Analysis of pol3-01 Yeast Mutants. Genes. 2023; 14(2):391. https://doi.org/10.3390/genes14020391
Chicago/Turabian StyleNir Heyman, Shaked, Mika Golan, Batia Liefshitz, and Martin Kupiec. 2023. "A Role for the Interactions between Polδ and PCNA Revealed by Analysis of pol3-01 Yeast Mutants" Genes 14, no. 2: 391. https://doi.org/10.3390/genes14020391