
Homologous Recombination and Repair Functions Required for Mutagenicity during Yeast Meiosis

Abstract
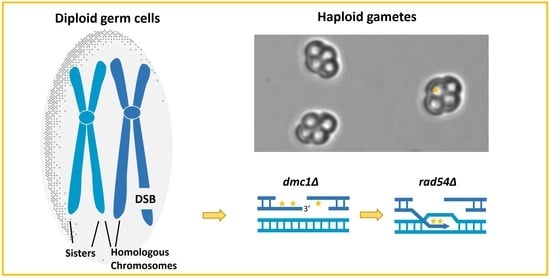
:
1. Introduction
2. Materials and Methods
2.1. Strain Construction
2.2. Media
2.3. Determining Mutation Rates during Mitotic Cell Divisions with Fluctuation Analysis
2.4. Determination of Meiotic Mutation and Recombination Rates
2.5. Meiotic Time Course Experiments
2.6. Meiotic Experiments Using the Inhibitor PP1
2.7. Statistical Treatments
3. Results
3.1. Meiotic Mutations Depend on the Generation of DSBs
3.2. Recombinases and Proteins Involved in Strand Exchange Affect Meiotic Mutagenicity
3.3. Evaluation of Mutation Occurrence during Meiotic S-Phase in rad54∆ Cells
3.4. The Involvement of Mus81 in Mutagenicity of Meiotic Joint Molecule Resolution
4. Discussion
4.1. Rad54 Restrains Mutagenicity during Meiosis and in Mitotically Cycling Cells
4.2. Single-Stranded DNA May Enhance Meiotic Mutagenicity
4.3. Is Meiotic Joint Molecule Resolution a Mutagenic Process?
4.4. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Magni, G.E.; Von Borstel, R.C. Different Rates of Spontaneous Mutation during Mitosis and Meiosis in Yeast. Genetics 1962, 47, 1097–1108. [Google Scholar] [CrossRef] [PubMed]
- Magni, G.E. Origin and Nature of Spontaneous Mutations in Meiotic Organisms. J. Cell. Physiol. 1964, 64, 65–71. [Google Scholar] [CrossRef]
- Kiktev, D.A.; Sheng, Z.; Lobachev, K.S.; Petes, T.D. GC content elevates mutation and recombination rates in the yeast Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 2018, 115, E7109–E7118. [Google Scholar] [CrossRef] [PubMed]
- Rattray, A.; Santoyo, G.; Shafer, B.; Strathern, J.N. Elevated mutation rate during meiosis in Saccharomyces cerevisiae. PLOS Genet. 2015, 11, e1004910. [Google Scholar] [CrossRef]
- Mansour, O.; Morciano, L.; Zion, K.; Elgrabli, R.; Zenvirth, D.; Simchen, G.; Arbel-Eden, A. Timing of appearance of new mutations during yeast meiosis and their association with recombination. Curr. Genet. 2020, 66, 577–592. [Google Scholar] [CrossRef] [PubMed]
- Arbeithuber, B.; Betancourt, A.J.; Ebner, T.; Tiemann-Boege, I. Crossovers are associated with mutation and biased gene conversion at recombination hotspots. Proc. Natl. Acad. Sci. USA. 2015, 112, 2109–2114. [Google Scholar] [CrossRef]
- Halldorsson, B.V.; Palsson, G.; Stefansson, O.A.; Jonsson, H.; Hardarson, M.T.; Eggertsson, H.P.; Gunnarsson, B.; Oddsson, A.; Halldorsson, G.H.; Zink, F.; et al. Characterizing mutagenic effects of recombination through a sequence-level genetic map. Science 2019, 363, eaau1043. [Google Scholar] [CrossRef]
- Magni, G.E. The Origin of Spontaneous Mutations during Meiosis. Proc. Natl. Acad. Sci. USA 1963, 50, 975–980. [Google Scholar] [CrossRef]
- Simchen, G.; Mansour, O.; Morciano, L.; Zenvirth, D.; Arbel-Eden, A. Mutagenicity in haploid yeast meiosis resulting from repair of DSBs by the sister chromatid. Curr. Genet. 2021, 67, 799–806. [Google Scholar] [CrossRef]
- Claeys Bouuaert, C.; Pu, S.; Wang, J.; Oger, C.; Daccache, D.; Xie, W.; Patel, D.J.; Keeney, S. DNA-driven condensation assembles the meiotic DNA break machinery. Nature 2021, 592, 144–149. [Google Scholar] [CrossRef]
- Székvölgyi, L.; Nicolas, A. From meiosis to postmeiotic events: Homologous recombination is obligatory but flexible. FEBS J. 2010, 277, 571–589. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Sasaki, M.; Kniewel, R.; Murakami, H.; Blitzblau, H.G.; Tischfield, S.E.; Zhu, X.; Neale, M.J.; Jasin, M.; Socci, N.D.; et al. A hierarchical combination of factors shapes the genome-wide topography of yeast meiotic recombination initiation. Cell 2011, 144, 719–731. [Google Scholar] [CrossRef]
- Lam, I.; Keeney, S. Mechanism and regulation of meiotic recombination initiation. Cold Spring Harb. Perspect. Biol. 2015, 7, a016634. [Google Scholar] [CrossRef]
- Mimitou, E.P.; Yamada, S.; Keeney, S. A global view of meiotic double-strand break end resection. Science 2017, 355, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Neale, M.J.; Keeney, S. Clarifying the mechanics of DNA strand exchange in meiotic recombination. Nature 2006, 442, 153–158. [Google Scholar] [CrossRef]
- Liu, Y.; Gaines, W.A.; Callender, T.; Busygina, V.; Oke, A.; Sung, P.; Fung, J.C.; Hollingsworth, N.M. Down-Regulation of Rad51 Activity during Meiosis in Yeast Prevents Competition with Dmc1 for Repair of Double-Strand Breaks. PLoS Genet. 2014, 10, e1004005. [Google Scholar] [CrossRef] [PubMed]
- Schwacha, A.; Kleckner, N. Interhomolog bias during meiotic recombination: Meiotic functions promote a highly differentiated interhomolog-only pathway. Cell 1997, 90, 1123–1135. [Google Scholar] [CrossRef] [PubMed]
- Cloud, V.; Chan, Y.-L.; Grubb, J.; Budke, B.; Bishop, D.K. Rad51 Is an Accessory factor for dmc1-mediated joint molecule formation during meiosis. Science 2012, 337, 1222–1225. [Google Scholar] [CrossRef] [PubMed]
- Mazin, A.V.; Mazina, O.M.; Bugreev, D.V.; Rossi, M.J. Rad54, the motor of homologous recombination. DNA Repair 2010, 9, 286–302. [Google Scholar] [CrossRef]
- Sanchez-Rebato, M.H.; Bouatta, A.M.; Gallego, M.E.; White, C.I.; Da Ines, O. RAD54 is essential for RAD51-mediated repair of meiotic DSB in Arabidopsis. PLoS Genet. 2021, 17, e1008919. [Google Scholar] [CrossRef]
- Arbel, A.; Zenvirth, D.; Simchen, G. Sister chromatid-based DNA repair is mediated by RAD54, not by DMC1 or TID1. Embo J. 1999, 18, 2648–2658. [Google Scholar] [CrossRef]
- Shinohara, M.; Shita-Yamaguchi, E.; Buerstedde, J.M.; Shinagawa, H.; Ogawa, H.; Shinohara, A. Characterization of the roles of the Saccharomyces cerevisiae RAD54 gene and a homologue of RAD54, RDH54/TID1, in mitosis and meiosis. Genetics 1997, 147, 1545–1556. [Google Scholar] [CrossRef] [PubMed]
- Nimonkar, A.V.; Dombrowski, C.C.; Siino, J.S.; Stasiak, A.Z.; Stasiak, A.; Kowalczykowski, S.C. Saccharomyces cerevisiae Dmc1 and Rad51 proteins preferentially function with Tid1 and Rad54 proteins, respectively, to promote DNA strand invasion during genetic recombination. J. Biol. Chem. 2012, 287, 28727–28737. [Google Scholar] [CrossRef]
- Holzen, T.M.; Shah, P.P.; Olivares, H.A.; Bishop, D.K. Tid1/Rdh54 promotes dissociation of Dmc1 from nonrecombinogenic sites on meiotic chromatin. Genes Dev. 2006, 20, 2593–2604. [Google Scholar] [CrossRef]
- Tsubouchi, H.; Roeder, G.S. Budding yeast Hed1 down-regulates the mitotic recombination machinery when meiotic recombination is impaired. Genes Dev. 2006, 20, 1766–1775. [Google Scholar] [CrossRef] [PubMed]
- Busygina, V.; Saro, D.; Williams, G.; Leung, W.K.; Say, A.F.; Sehorn, M.G.; Sung, P.; Tsubouchi, H. Novel Attributes of Hed1 Affect Dynamics and Activity of the Rad51 Presynaptic Filament during Meiotic Recombination. J. Biol. Chem. 2012, 287, 1566–1575. [Google Scholar] [CrossRef] [PubMed]
- Crickard, J.B.; Kaniecki, K.; Kwon, Y.; Sung, P.; Lisby, M.; Greene, E.C. Regulation of Hed1 and Rad54 binding during maturation of the meiosis-specific presynaptic complex. EMBO J. 2018, 37, e98728. [Google Scholar] [CrossRef]
- Niu, H.; Wan, L.; Busygina, V.; Kwon, Y.; Allen, J.A.; Li, X.; Kunz, R.C.; Kubota, K.; Wang, B.; Sung, P.; et al. Regulation of Meiotic Recombination via Mek1-Mediated Rad54 Phosphorylation. Mol. Cell 2009, 36, 393–404. [Google Scholar] [CrossRef]
- Hollingsworth, N.M. Phosphorylation and the creation of interhomolog bias during meiosis in yeast. Cell Cycle 2010, 9, 436–437. [Google Scholar] [CrossRef]
- Busygina, V.; Sehorn, M.G.; Shi, I.Y.; Tsubouchi, H.; Roeder, G.S.; Sung, P. Hed1 regulates Rad51-mediated recombination via a novel mechanism. Genes Dev. 2008, 22, 786–795. [Google Scholar] [CrossRef]
- Cao, L.; Alani, E.; Kleckner, N. A pathway for generation and processing of double-strand breaks during meiotic recombination in S. cerevisiae. Cell 1990, 61, 1089–1101. [Google Scholar] [CrossRef] [PubMed]
- Kassir, Y.; Simchen, G. Monitoring meiosis and sporulation in Saccharomyces cerevisiae. Methods Enzymol. 1991, 194, 94–110. [Google Scholar] [PubMed]
- Rose, M.; Winston, F.; Hieter, P. Methods Yeast Genetics—A Laboratory Course Manual; Cold Spring Harbor: New York, NY, USA, 1990. [Google Scholar]
- I Lang, G.; Murray, A.W. Estimating the per-base-pair mutation rate in the yeast Saccharomyces cerevisiae. Genetics 2008, 178, 67–82. [Google Scholar] [CrossRef] [PubMed]
- Luria, S.E.; Delbrück, M. Mutations of Bacteria from Virus Sensitivity to Virus Resistance. Genetics 1943, 28, 491–511. [Google Scholar] [CrossRef] [PubMed]
- Rosche, W.A.; Foster, P.L. Determining mutation rates in bacterial populations. Methods 2000, 20, 4–17. [Google Scholar] [CrossRef]
- Arbel-Eden, A.; Joseph-Strauss, D.; Masika, H.; Printzental, O.; Rachi, E.; Simchen, G. Trans-Lesion DNA Polymerases May Be Involved in Yeast Meiosis. G3 Genes Genomes Genet. 2013, 3, 633–644. [Google Scholar] [CrossRef]
- Zenvirth, D.; Loidl, J.; Klein, S.; Arbel, A.; Shemesh, R.; Simchen, G. Switching yeast from meiosis to mitosis: Double-strand break repair, recombination and synaptonemal complex. Genes Cells 1997, 2, 487–498. [Google Scholar] [CrossRef]
- Bishop, D.K.; Park, D.; Xu, L.; Kleckner, N. DMC1: A meiosis-specific yeast homolog of E. coli recA required for recombination, synaptonemal complex formation, and cell cycle progression. Cell 1992, 69, 439–456. [Google Scholar] [CrossRef]
- Wan, L.; Zhang, C.; Shokat, K.M.; Hollingsworth, N.M. Chemical inactivation of cdc7 kinase in budding yeast results in a reversible arrest that allows efficient cell synchronization prior to meiotic recombination. Genetics 2006, 174, 1767–1774. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Ng, A.W.T.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef]
- Keeney, S. Mechanism and control of meiotic recombination initiation. Curr. Top. Dev. Biol. 2001, 52, 1–53. [Google Scholar]
- Brown, M.S.; Bishop, D.K. DNA strand exchange and RecA homologs in meiosis. Cold Spring Harb. Perspect. Biol. 2014, 7, a016659. [Google Scholar] [CrossRef]
- Shinohara, A.; Ogawa, H.; Ogawa, T. Rad51 protein involved in repair and recombination in S. cerevisiae is a RecA-like protein. Cell 1992, 69, 457–470. [Google Scholar] [CrossRef]
- Lao, J.P.; Cloud, V.; Huang, C.-C.; Grubb, J.; Thacker, D.; Lee, C.-Y.; Dresser, M.E.; Hunter, N.; Bishop, D.K. Meiotic Crossover Control by Concerted Action of Rad51-Dmc1 in Homolog Template Bias and Robust Homeostatic Regulation. PLoS Genet. 2013, 9, e1003978. [Google Scholar] [CrossRef] [PubMed]
- Ziesel, A.; Weng, Q.; Ahuja, J.S.; Bhattacharya, A.; Dutta, R.; Cheng, E.; Börner, G.V.; Lichten, M.; Hollingsworth, N.M. Rad51-mediated interhomolog recombination during budding yeast meiosis is promoted by the meiotic recombination checkpoint and the conserved Pif1 helicase. PLoS Genet. 2022, 18, e1010407. [Google Scholar] [CrossRef]
- Klein, H.L. RDH54, a RAD54 homologue in Saccharomyces cerevisiae, is required for mitotic diploid-specific recombination and repair and for meiosis. Genetics 1997, 147, 1533–1543. [Google Scholar] [CrossRef] [PubMed]
- Alexeev, A.; Mazin, A.; Kowalczykowski, S.C. Rad54 protein possesses chromatin-remodeling activity stimulated by the Rad51–ssDNA nucleoprotein filament. Nat. Struct. Biol. 2003, 10, 182–186. [Google Scholar] [CrossRef]
- Prasad, T.K.; Robertson, R.B.; Visnapuu, M.L.; Chi, P.; Sung, P.; Greene, E.C. A DNA-translocating Snf2 Molecular Motor: Saccharomyces cerevisiae Rdh54 Displays Processive Translocation and Extrudes DNA Loops. J. Mol. Biol. 2007, 369, 940–953. [Google Scholar] [CrossRef] [PubMed]
- Jessop, L.; Lichten, M. Mus81/Mms4 Endonuclease and Sgs1 Helicase Collaborate to Ensure Proper Recombination Intermediate Metabolism during Meiosis. Mol. Cell. 2008, 31, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.D.; Lao, J.P.; Taylor, A.F.; Smith, G.R.; Hunter, N. RecQ Helicase, Sgs1, and XPF Family Endonuclease, Mus81-Mms4, Resolve Aberrant Joint Molecules during Meiotic Recombination. Mol. Cell 2008, 31, 324–336. [Google Scholar] [CrossRef]
- Bergero, R.; Ellis, P.; Haerty, W.; Larcombe, L.; Macaulay, I.; Mehta, T.; Mogensen, M.; Murray, D.; Nash, W.; Neale, M.J.; et al. Meiosis and beyond—Understanding the mechanistic and evolutionary processes shaping the germline genome. Biol. Rev. 2021, 96, 822–841. [Google Scholar] [CrossRef]
- Arbel-Eden, A.; Simchen, G. Elevated Mutagenicity in Meiosis and Its Mechanism. BioEssays 2019, 41, 272–281. [Google Scholar] [CrossRef]
- Bishop, D.K. Rad51, the lead in mitotic recombinational DNA repair, plays a supporting role in budding yeast meiosis. Cell Cycle 2012, 11, 4105–4106. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, M.; Gasior, S.L.; Bishop, D.K.; Shinohara, A. Tid1/Rdh54 promotes colocalization of rad51 and dmc1 during meiotic recombination. Proc. Natl. Acad. Sci. USA 2000, 97, 10814–10819. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, M.; Sakai, K.; Shinohara, A.; Bishop, D.K. Crossover interference in Saccharomyces cerevisiae requires a T1D1/RDH54- and DMC1-dependent pathway. Genetics 2003, 163, 1273–1286. [Google Scholar] [CrossRef] [PubMed]
- Tsubouchi, H.; Roeder, G. The importance of genetic recombination for fidelity of chromosome pairing in meiosis. Dev. Cell 2003, 5, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Callender, T.L.; Laureau, R.; Wan, L.; Chen, X.; Sandhu, R.; Laljee, S.; Zhou, S.; Suhandynata, R.T.; Prugar, E.; Gaines, W.A.; et al. Mek1 Down Regulates Rad51 Activity during Yeast Meiosis by Phosphorylation of Hed1. PLoS Genet. 2016, 12, e1006226. [Google Scholar] [CrossRef]
- Johnson, R.; Borde, V.; Neale, M.J.; Bishop-Bailey, A.; North, M.; Harris, S.; Nicolas, A.; Goldman, A.S.H. Excess single-stranded DNA inhibits meiotic double-strand break repair. PLoS Genet. 2007, 3, e223. [Google Scholar] [CrossRef] [PubMed]
- Hong, E.L.; Shinohara, A.; Bishop, D.K. Saccharomyces cerevisiae Dmc1 Protein Promotes Renaturation of Single-strand DNA (ssDNA) and Assimilation of ssDNA into Homologous Super-coiled Duplex DNA. J. Biol. Chem. 2001, 276, 41906–41912. [Google Scholar] [CrossRef]
- MacQueen, A.J. Catching a (Double-Strand) Break: The Rad51 and Dmc1 Strand Exchange Proteins Can Co-occupy Both Ends of a Meiotic DNA Double-Strand Break. PLoS Genet. 2015, 11, e1005741. [Google Scholar] [CrossRef]
- Lindahl, T. Instability and decay of the primary structure of DNA. Nature 1993, 362, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Roberts, S.A.; Sterling, J.; Thompson, C.; Harris, S.; Mav, D.; Shah, R.; Klimczak, L.J.; Kryukov, G.V.; Malc, E.; Mieczkowski, P.A.; et al. Clustered Mutations in Yeast and in Human Cancers Can Arise from Damaged Long Single-Strand DNA Regions. Mol. Cell 2012, 46, 424–435. [Google Scholar] [CrossRef]
- Saini, N.; Gordenin, D.A. Hypermutation in single-stranded DNA. DNA Repair 2020, 91–92, 102868. [Google Scholar] [CrossRef]
- Hollingsworth, N.M.; Brill, S.J. The Mus81 solution to resolution: Generating meiotic crossovers without Holliday junctions. J. Bone Jt. Surg. 2004, 18, 117–125. [Google Scholar] [CrossRef] [PubMed]
- De los Santos, T.; Hunter, N.; Lee, C.; Larkin, B.; Loidl, J.; Hollingsworth, N.M. The MUS81/MMS4 endonuclease acts independently of double-holliday junction resolution to promote a distinct subset of crossovers during meiosis in budding yeast. Genetics 2003, 164, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Sanchez, A.; Adam, C.; Ranjha, L.; Reginato, G.; Chervy, P.; Tellier-Lebegue, C.; Andreani, J.; Guérois, R.; Ropars, V.; et al. Molecular basis of the dual role of the Mlh1-Mlh3 endonuclease in MMR and in meiotic crossover formation. Proc. Natl. Acad. Sci. USA 2021, 118, e2022704118. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain Name | Relevant Mutations | Genotype |
|---|---|---|
| YRA10 | WT, MATa | MATa, ho∆::LYS2, lys2, leu2, his4X, trp1∆::hisG, ura3, ade2∆::hisG, ADE2-CAN1 (ADE2 inserted upstream of CAN1), TRP1-LYP1 (TRP1 inserted upstream of LYP1) |
| YRA12 | WT, MATα | MATα, ho∆::LYS2, lys2, leu2, his4B::LEU2, trp1∆::hisG, ura3, ade2∆::hisG, avt2∆::URA3 (URA3 inserted downstream of can1), can1::kanMX6, lyp1::natNT2 |
| YRA23-1 | diploid WT, CDC7 | YRA10 × YRA12 |
| YRA17 | spo11∆ | YRA23-1 with homozygous spo11∆::hisG |
| YRA34-1 | mre11∆ | YRA23-1 with homozygous mre11∆::kanMX4 |
| YRA20 | dmc1∆ | YRA23-1 with homozygous dmc1∆::kanMX4 |
| YRA55 | hed1∆ | YRA23-1 with homozygous hed1∆::kanMX4 (ydr015c) |
| YRA56 | hed1∆ dmc1∆ | YRA23-1 with homozygous hed1∆::kanMX4 (ydr015c), dmc1∆::kanMX4 |
| #3512 | rad54∆, MATa | MATa, ho∆::LYS2, lys2, ura3, leu2∆::hisG, his4X::LEU2-BamHI-URA3, rad54∆::URA3 |
| ALY77 | rad54∆, MATa | MATa, ho∆::LYS2, lys2, leu2, his4X, trp1∆::hisG, ura3, ade2:∆::kanMX4, ADE2-CAN1 (ADE2 inserted upstream of CAN1), TRP1-LYP1 (TRP1 inserted upstream of LYP1), rad54∆::URA3 |
| ALY92 | rad54∆, MATα | MATα, ho∆::LYS2, lys2, leu2, his4B::LEU2, trp1∆::hisG, ura3, ade2∆::kanMX4, avt2∆::URA3 (URA3 inserted downstream of can1), can1::kanMX6, lyp1::natNT2, rad54∆::URA3 |
| ALY94 | diploid, rad54∆ | ALY77 × ALY92 |
| YAS3 | tid1∆ | YRA23-1 with homozygous tid1∆::kanMX4 |
| YRA51 | mus81∆ | YRA23-1 with homozygous mus81∆::kanMX4 |
| OMY78-1 | WT, MATa, cdc7-as3-9myc | MATa, hoΔ::LYS2, leu2Δ::hisG, his4x, lys2, trp1∆::hygB, ura3∆::hisG, ade2∆::kanMX4, cdc7-as3-9myc, ADE2-CAN1 (ADE2 inserted upstream of CAN1), TRP1-LYP1 (TRP1 inserted upstream of LYP1) |
| OMY75-2 | WT, MATα, cdc7-as3-9myc | MATα, ho∆::LYS2, ade2∆::kanMX4, trp1∆::hph, his4B::LEU2, avt2∆::URA3 (URA3 inserted downstream of can1), cdc7-as3-9myc, can1::kanMX6, lyp1::natNT2 |
| OMY93 | diploid WT, cdc7-ac3-9myc | OMY78-1 × OMY75-2 |
| ALY170 | rad54∆, cdc7-as3-9myc | ALY94 with homozygous cdc7-as3-9myc |
| Mutated Gene | % Cell Viability (at 8 h) | Spore Germination | Recombination at HIS4 (at 8 h) (×10−2) | Mutation at CAN1 (8 h) | |
|---|---|---|---|---|---|
| Meiotic Mutations (×10−6) | Fold Change Mutant/ WT | ||||
| WT | 100 | 96% (366/380) | 1.87 | 1.20 | 1.00 |
| spo11∆ | 36 | 0% (0/150) | 0.00 | 0.12 | 0.10 |
| mre11∆ | 56 | 0% (0/146) | 0.01 | 0.24 | 0.20 |
| dmc1∆ | 108 | Φ | 0.81 | 4.33 | 3.60 |
| hed1∆ | 96 | 93% (164/176) | 0.91 | 0.39 | 0.33 |
| hed1∆dmc1∆ | 93 | 55% (173/312) | 0.22 | 0.18 | 0.15 |
| rad54∆ | 84 | 66% (105/160) | 1.97 * | 9.70 | 8.07 |
| tid1∆ | 58 | N.D | 0.65 | 0.11 | 0.09 |
| mus81∆ | 58 | 56% (158/280) | 0.71 | 0.85 * | 0.71 |
| Mutated Gene | Strain | Recombinants around CAN1 Reporter in CANR Mutants | Fold Change Mutant/WT |
|---|---|---|---|
| WT | YRA23-1 | 39% (95/243) | 1 |
| hed1∆ | YRA55 | 40% (69/171) | 1 |
| hed1∆dmc1∆ | YRA56 | 25% (38/153) | 0.64 |
| mus81∆ | YRA51 | 15% (34/229) | 0.38 |
| Mutated Gene | Strain | Mitotic Mutation Rate at CAN1 (×10−6) In Mitotic Cells | Fold Change Mutant/WT |
|---|---|---|---|
| WT | YRA10 | 0.2 | 1 |
| rad54∆ | ALY94 | 5.14 | 25.7 |
| tid1∆ | YAS3 | 0.3 | 1.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morciano, L.; Elgrabli, R.M.; Zenvirth, D.; Arbel-Eden, A. Homologous Recombination and Repair Functions Required for Mutagenicity during Yeast Meiosis. Genes 2023, 14, 2017. https://doi.org/10.3390/genes14112017
Morciano L, Elgrabli RM, Zenvirth D, Arbel-Eden A. Homologous Recombination and Repair Functions Required for Mutagenicity during Yeast Meiosis. Genes. 2023; 14(11):2017. https://doi.org/10.3390/genes14112017
Chicago/Turabian StyleMorciano, Liat, Renana M. Elgrabli, Drora Zenvirth, and Ayelet Arbel-Eden. 2023. "Homologous Recombination and Repair Functions Required for Mutagenicity during Yeast Meiosis" Genes 14, no. 11: 2017. https://doi.org/10.3390/genes14112017