
Characterizing the Complete Mitochondrial Genomes of Three Bugs (Hemiptera: Heteroptera) Harming Bamboo

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Observation of the Damaged Condition and Occurrence Regularity
2.2. Sample Isolation and DNA Extraction
2.3. Genome Assembly, Annotation, and Analysis
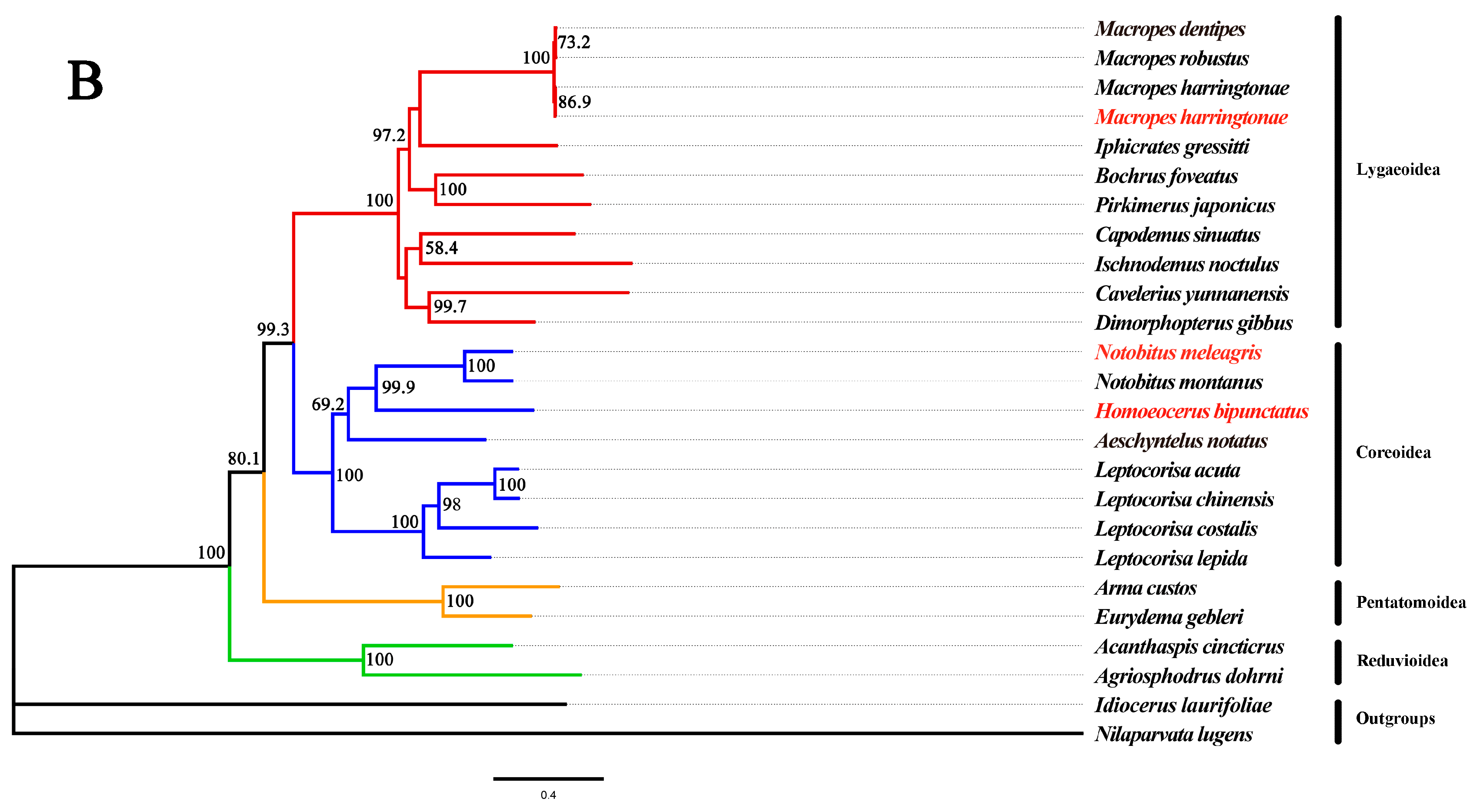
2.4. Phylogenetic Analysis
3. Results
3.1. Hazard Condition and Occurrence Regularity
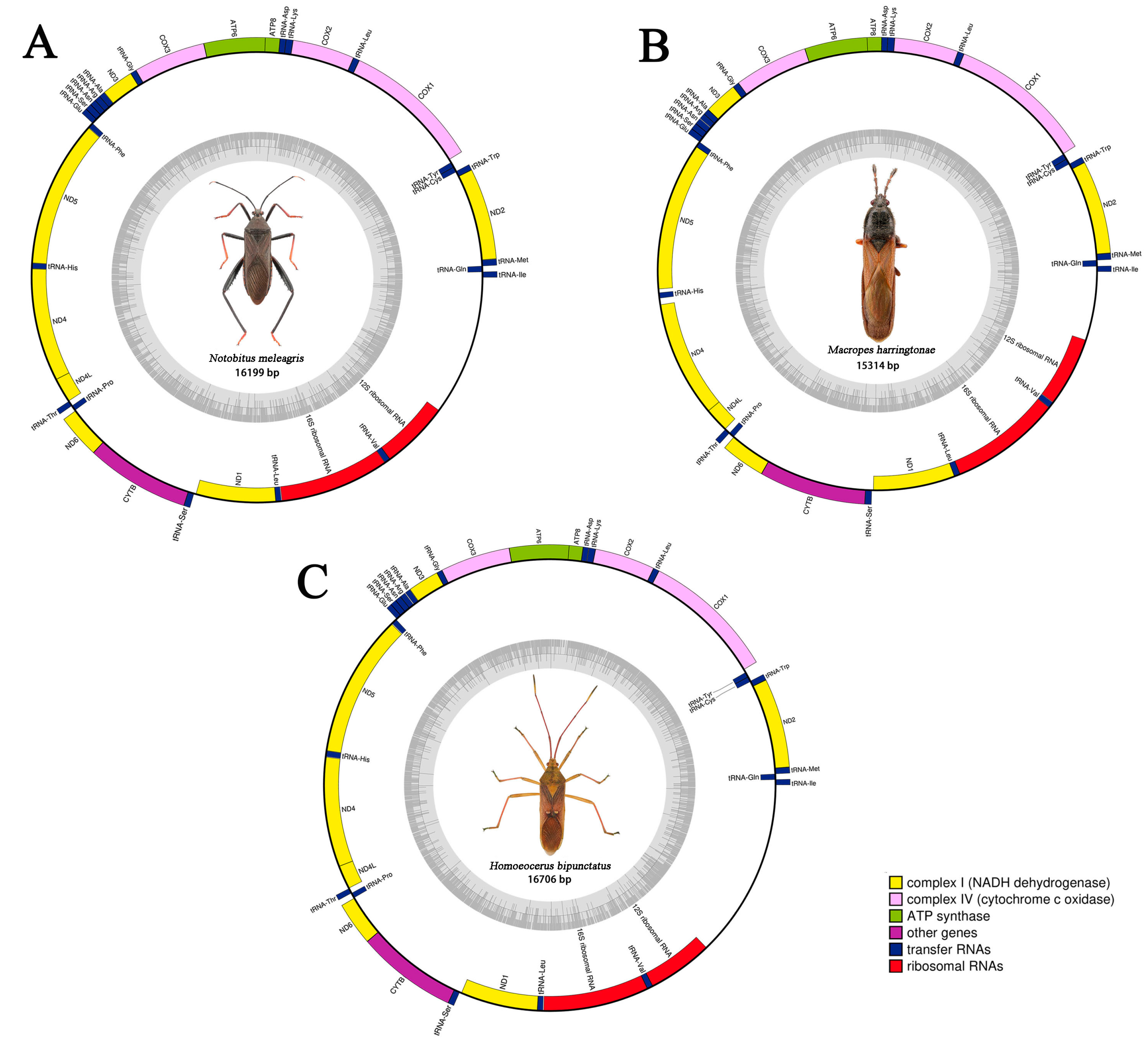
3.2. Mitogenomic Organization and Composition
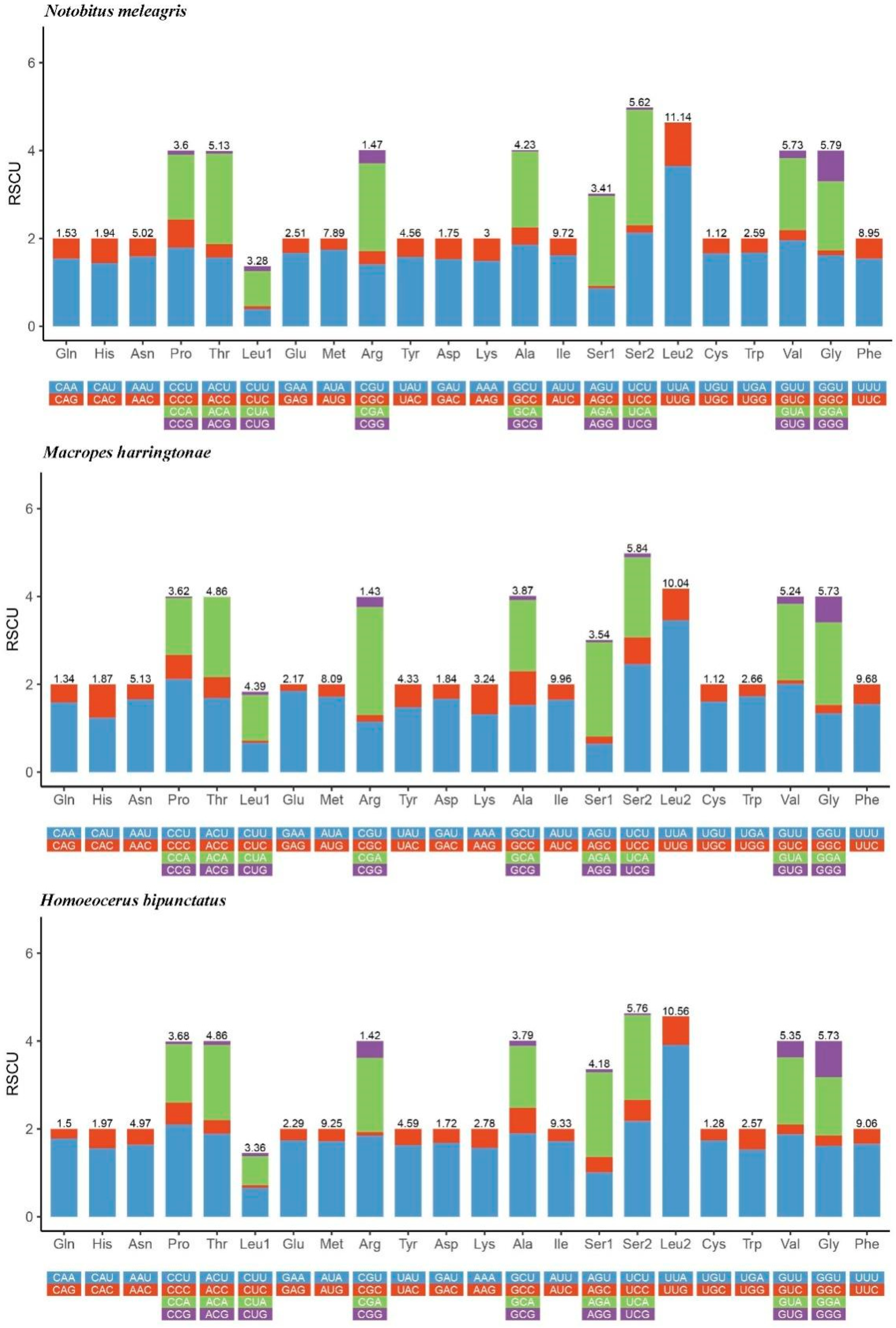
3.3. PCGs and Codon Usage
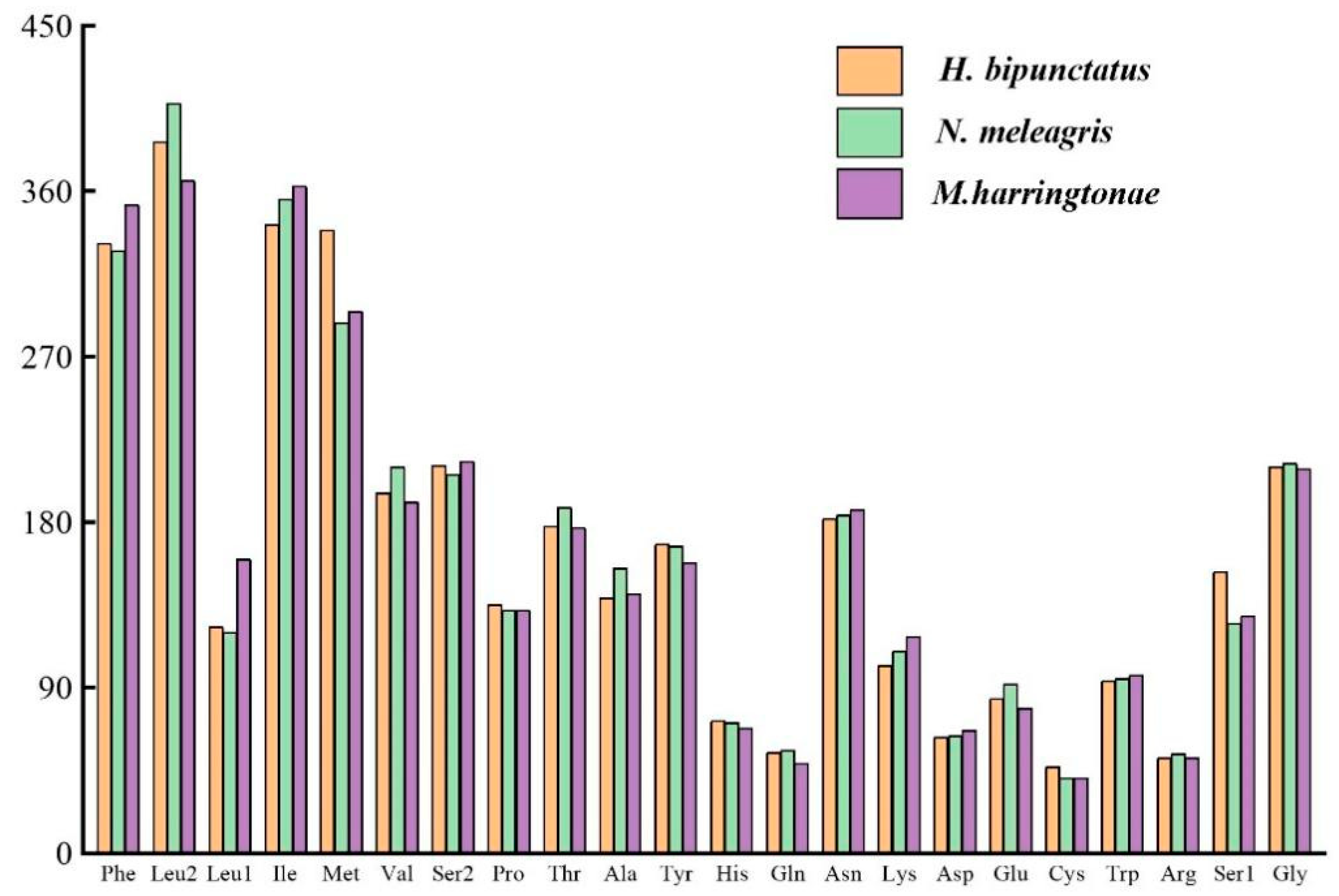
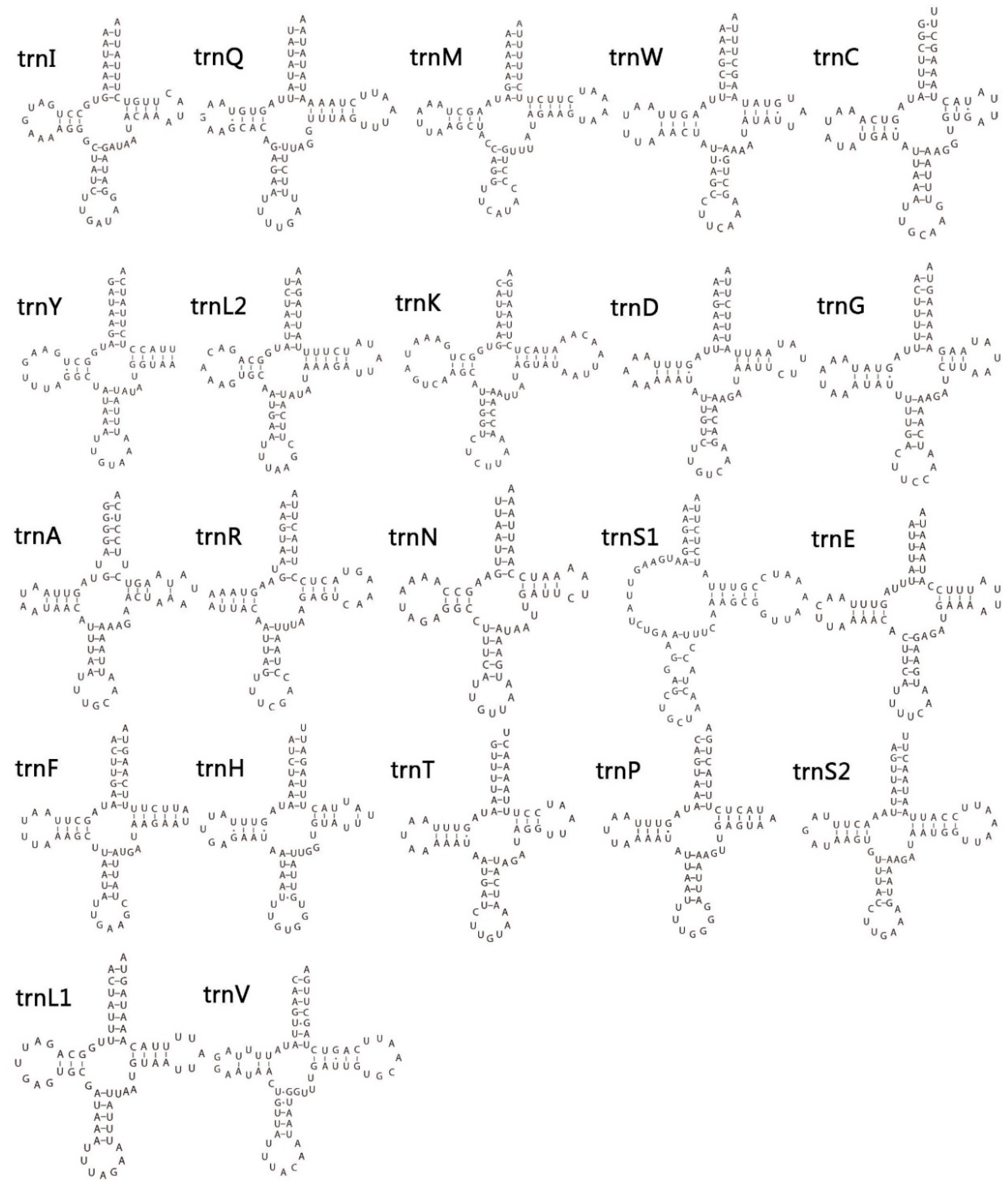
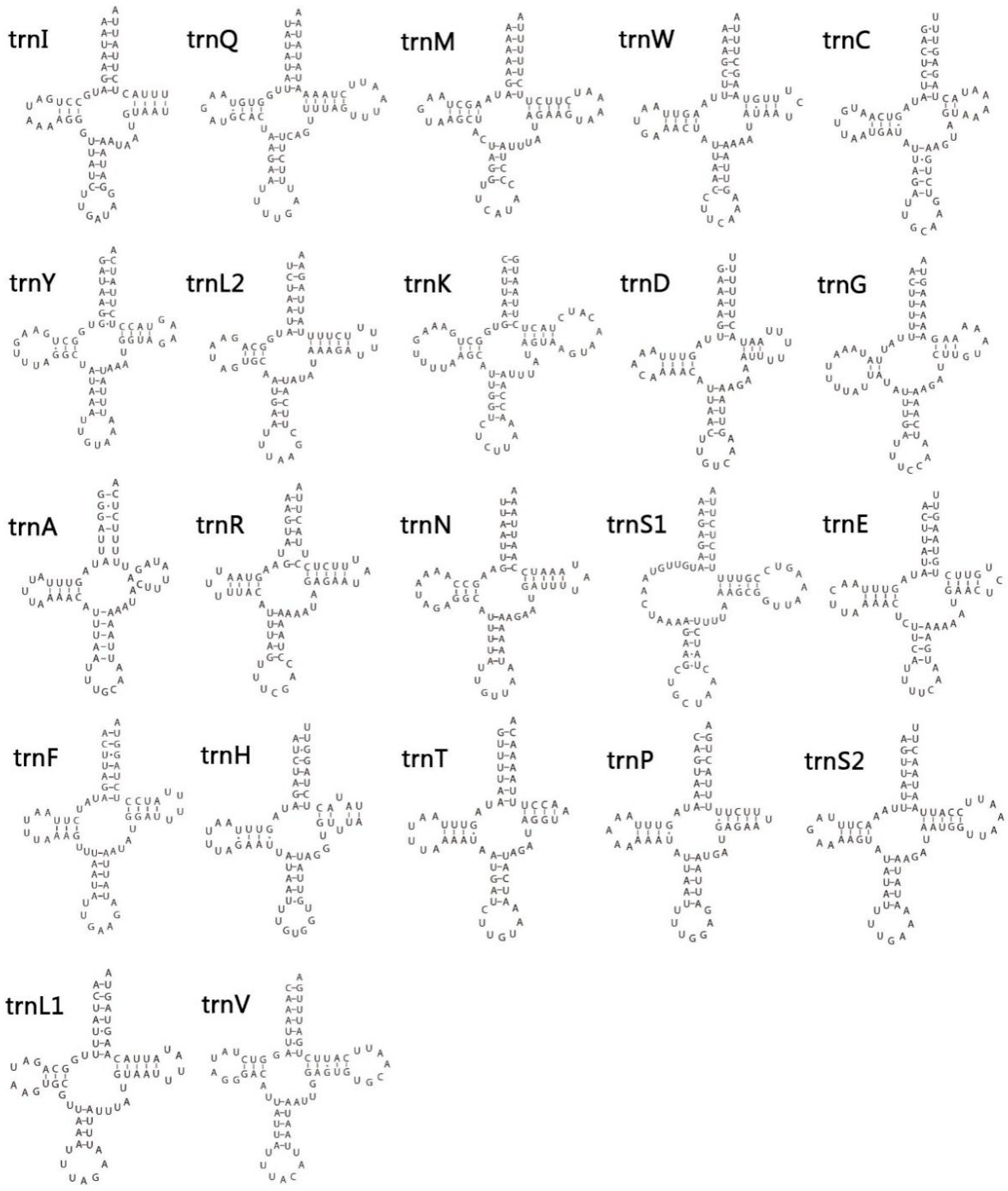
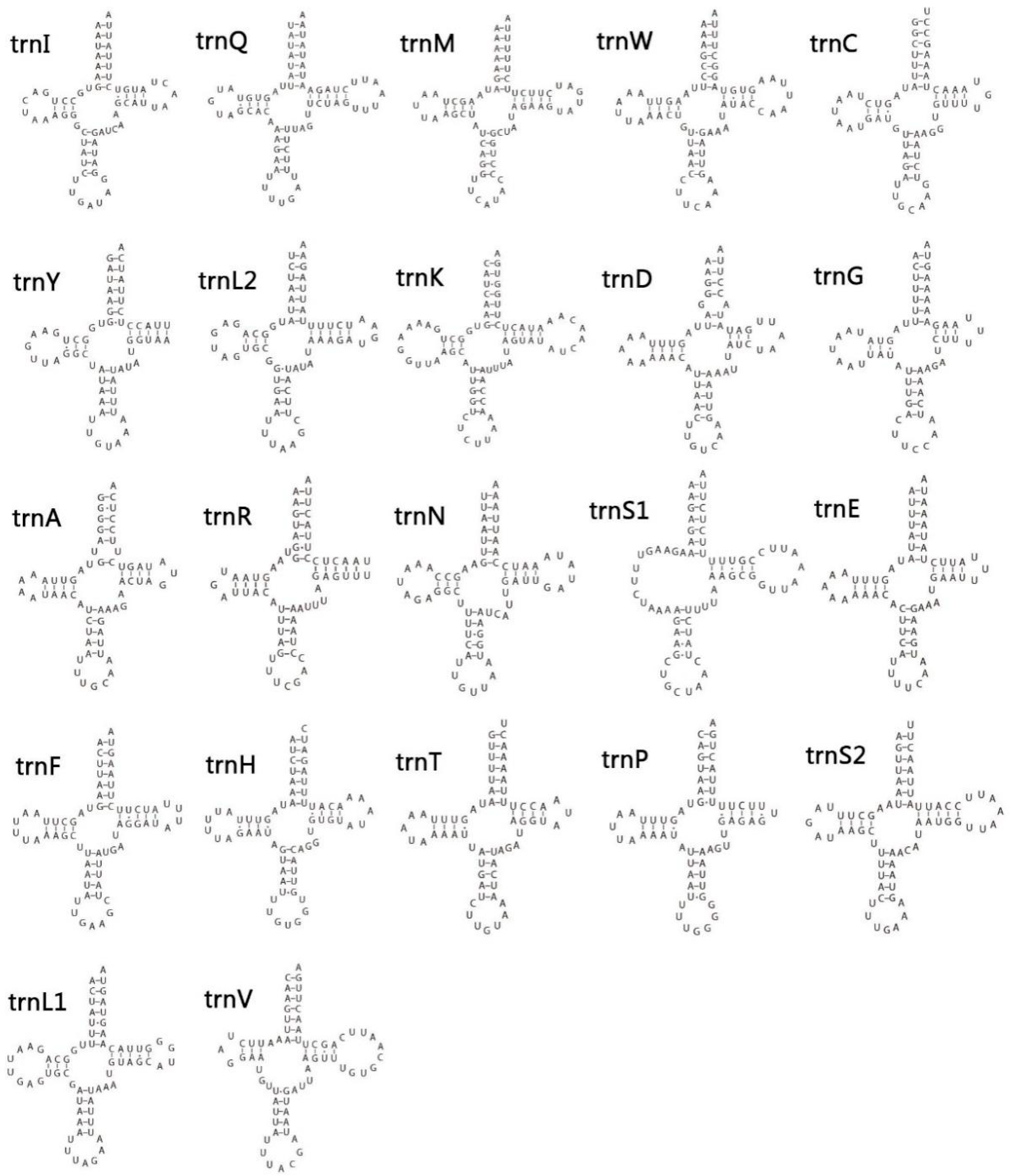
3.4. Transfer and Ribosomal RNA Genes
3.5. Control Region
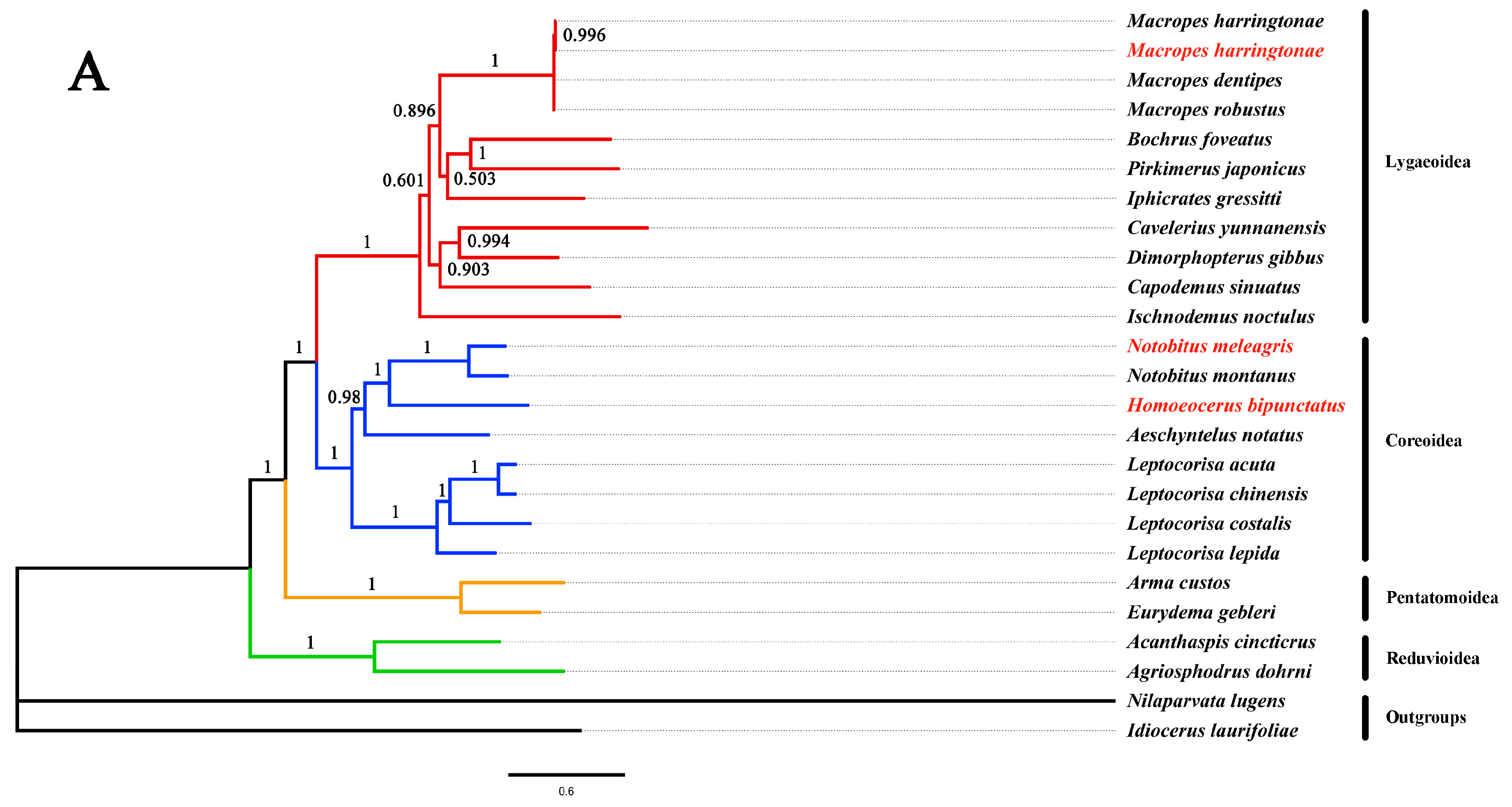
3.6. Phylogenetic Analyses
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Superfamily | Species | NCBI No. | Length | |
|---|---|---|---|---|
| Ingroups | Lygaeoidea | M. harringtonae | NC065820 | 14,942 |
| Lygaeoidea | M. harringtonae | OP442511 | 15,314 | |
| Lygaeoidea | Macropes dentipes | NC065821 | 14,923 | |
| Lygaeoidea | Macropes robustus | NC065822 | 15,041 | |
| Lygaeoidea | Bochrus foveatus | NC065814 | 14,738 | |
| Lygaeoidea | P. japonicus | NC065823 | 15,440 | |
| Lygaeoidea | Iphicrates gressitti | NC065818 | 15,288 | |
| Lygaeoidea | Cavelerius yunnanensis | NC065816 | 15,330 | |
| Lygaeoidea | Dimorphopterus gibbus | NC065817 | 14,988 | |
| Lygaeoidea | Capodemus sinuatus | NC065815 | 15,199 | |
| Lygaeoidea | Ischnodemus noctulus | NC065819 | 15,291 | |
| Coreoidea | N. meleagris | OP442510 | 16,199 | |
| Coreoidea | Notobitus montanus | NC065112 | 16,209 | |
| Coreoidea | H. bipunctatus | OP442512 | 16,706 | |
| Coreoidea | A. notatus | NC012446 | 14,532 | |
| Coreoidea | Leptocorisa acuta | NC061738 | 15,373 | |
| Coreoidea | Leptocorisa chinensis | NC061737 | 15,433 | |
| Coreoidea | Leptocorisa costalis | NC061680 | 15,353 | |
| Coreoidea | Leptocorisa lepida | NC061739 | 15,129 | |
| Pentatomoidea | Arma custos | NC051562 | 15,629 | |
| Pentatomoidea | Eurydema gebleri | NC027489 | 16,005 | |
| Reduvioidea | Acanthaspis cincticrus | NC037735 | 15,686 | |
| Reduvioidea | Agriosphodrus dohrni | NC015842 | 16,470 | |
| Outgroups | Fulgoroidea | N. lugens | NC021748 | 17,619 |
| Membracoidea | I. laurifoliae | NC039741 | 16,811 |
| M. harringtonae | ||||||||
| Generation | Mouth | |||||||
| Mar. | Apr. | May | Jun. | Jul. | Aug. | Sep. | Oct.−Feb. | |
| Overwintering generation | (−) | − | − | |||||
| (+) | + | + | ||||||
| • | • | • | ||||||
| First generation | − | − | − | |||||
| + | + | + | ||||||
| • | • | • | ||||||
| Second generation | − | − | − | |||||
| + | + | + | ||||||
| • | • | • | ||||||
| Third generation | − | − | − | (−) | ||||
| + | (+) | |||||||
| H. bipunctatus | ||||||||
| Generation | Mouth | |||||||
| Apr. | May | Jun. | Jul. | Aug. | Sep. | Oct. | Nov.−Mar. | |
| Overwintering generation | (+) | + | + | + | ||||
| • | • | • | ||||||
| First generation | − | − | − | |||||
| + | + | + | + | |||||
| • | • | • | • | |||||
| Second generation | − | − | − | − | ||||
| + | + | + | (+) | |||||
| N. meleagris | ||||||
| Gene | Direction | Location | Size (bp) | Start Codon | Stop Codon | INC |
| trnI | J | 1–65 | 65 | - | - | 0 |
| trnQ | N | 63–131 | 69 | - | - | −3 |
| trnM | J | 131–199 | 69 | - | - | −1 |
| nad2 | J | 201–1200 | 1000 | ATG | T | 1 |
| trnW | J | 1201–1264 | 64 | - | - | 0 |
| trnC | N | 1257–1319 | 63 | - | - | 6 |
| trnY | N | 1320–1382 | 63 | - | - | 0 |
| cox1 | J | 1420–2917 | 1498 | ATT | T | 37 |
| trnL2 | J | 2918–2984 | 67 | - | - | 0 |
| cox2 | J | 2985–3663 | 679 | ATC | T | 0 |
| trnK | J | 3663–3738 | 75 | - | - | −1 |
| trnD | J | 3739–3803 | 65 | - | - | 0 |
| atp8 | J | 3804–3965 | 162 | ATA | TAA | 0 |
| atp6 | J | 3959–4630 | 672 | ATG | TAA | −7 |
| cox3 | J | 4630–5416 | 787 | ATG | T | −1 |
| trnG | J | 5417–5480 | 64 | - | - | 0 |
| nad3 | J | 5481–5832 | 352 | ATA | T | 0 |
| trnA | J | 5834–5897 | 64 | - | - | 1 |
| trnR | J | 5898–5961 | 64 | - | - | 0 |
| trnN | J | 5962–6027 | 66 | - | - | 0 |
| trnS1 | J | 6027–6096 | 70 | - | - | −1 |
| trnE | J | 6096–6160 | 65 | - | - | −1 |
| trnF | N | 6161–6225 | 65 | - | - | 0 |
| nad5 | N | 6228–7938 | 1711 | ATG | T | 2 |
| trnH | N | 7940–8002 | 63 | - | - | 1 |
| nad4 | N | 8004–9318 | 1315 | ATG | T | 1 |
| nad4L | N | 9314–9602 | 289 | ATT | T | −5 |
| trnT | J | 9605–9667 | 63 | - | - | 2 |
| trnP | N | 9668–9730 | 63 | - | - | 0 |
| nad6 | J | 9736–10,218 | 483 | ATC | TAA | 5 |
| cytb | J | 10,218–11,352 | 1135 | ATG | T | −1 |
| trnS2 | J | 11,353–11,421 | 69 | - | - | 0 |
| nad1 | N | 11,443–12,367 | 925 | ATT | T | 21 |
| trnL1 | N | 12,368–12,432 | 65 | - | - | 0 |
| rrnL | N | 12,437–13,707 | 1271 | - | - | 4 |
| trnV | N | 13,709–13,776 | 68 | - | - | 1 |
| rrnS | N | 13,777–14,572 | 796 | - | - | 0 |
| D-−loop | J | 14,573–16,199 | 1627 | - | - | 0 |
| M. harringtonae | ||||||
| Gene | Direction | Location | Size (bp) | Start codon | Stop codon | INC |
| trnI | J | 1–62 | 62 | - | - | 0 |
| trnQ | N | 60–128 | 69 | - | - | −3 |
| trnM | J | 128–195 | 68 | - | - | −1 |
| nad2 | J | 196–1186 | 991 | ATT | T | 0 |
| trnW | J | 1187–1249 | 63 | - | - | 0 |
| trnC | N | 1249–1312 | 64 | - | - | −1 |
| trnY | N | 1313–1377 | 65 | - | - | 0 |
| cox1 | J | 1380–2913 | 1543 | TTG | T | 2 |
| trnL2 | J | 2914–2978 | 65 | - | - | 0 |
| cox2 | J | 2979–3657 | 679 | ATA | T | 0 |
| trnK | J | 3658–3729 | 72 | - | - | 0 |
| trnD | J | 3730–3792 | 63 | - | - | 0 |
| atp8 | J | 3793–3948 | 156 | ATT | TAA | 0 |
| atp6 | J | 3942–4607 | 666 | ATG | TAA | −7 |
| cox3 | J | 4607–5390 | 784 | ATG | T | −1 |
| trnG | J | 5391–5456 | 66 | - | - | 0 |
| nad3 | J | 5457–5810 | 354 | ATT | TAG | 0 |
| trnA | J | 5810–5871 | 62 | - | - | −1 |
| trnR | J | 5872–5935 | 64 | - | - | 0 |
| trnN | J | 5939–6005 | 66 | - | - | 3 |
| trnS1 | J | 6005–6073 | 69 | - | - | −1 |
| trnE | J | 6073–6136 | 64 | - | - | −1 |
| trnF | N | 6137–6201 | 65 | - | - | 0 |
| nad5 | N | 6199–7870 | 1672 | ATT | T | −3 |
| trnH | N | 7910–7972 | 63 | - | - | 39 |
| nad4 | N | 8044–9358 | 1315 | ATG | T | 71 |
| nad4L | N | 9352–9630 | 279 | ATT | TAA | −7 |
| trnT | J | 9633–9694 | 62 | - | - | 2 |
| trnP | N | 9695–9757 | 63 | - | - | 0 |
| nad6 | J | 9759–10,232 | 474 | ATA | TAA | 1 |
| cytb | J | 10,232–11,363 | 1132 | ATG | T | −1 |
| trnS2 | J | 11,364–11,433 | 70 | - | - | 0 |
| nad1 | N | 11,450–12,370 | 921 | ATA | T | 16 |
| trnL1 | N | 12,371–12,435 | 65 | - | - | 0 |
| rrnL | N | 12,436–13,684 | 1249 | - | - | 4 |
| trnV | N | 13,686–13,753 | 68 | - | - | 1 |
| rrnS | N | 13,756–14,542 | 787 | - | - | 2 |
| D-−loop | J | 14,543–15,314 | 772 | - | - | 0 |
| H. bipunctatus | ||||||
| Gene | Direction | Location | Size (bp) | Start codon | Stop codon | INC |
| trnI | J | 1–65 | 65 | - | - | 0 |
| trnQ | N | 63–131 | 69 | - | - | −3 |
| trnM | J | 131–198 | 68 | - | - | −1 |
| nad2 | J | 199–1201 | 1003 | ATG | T | 0 |
| trnW | J | 1202–1269 | 68 | - | - | 0 |
| trnC | N | 1262–1324 | 63 | - | - | −8 |
| trnY | N | 1325–1386 | 62 | - | - | 0 |
| cox1 | J | 1424–2921 | 1498 | ATT | T | 37 |
| trnL2 | J | 2922–2986 | 65 | - | - | 0 |
| cox2 | J | 2987–3665 | 679 | ATC | T | 0 |
| trnK | J | 3666–3739 | 74 | - | - | 0 |
| trnD | J | 3741–3803 | 63 | - | - | 1 |
| atp8 | J | 3804–3962 | 159 | ATC | TAA | 0 |
| atp6 | J | 3956–4627 | 672 | ATG | TAA | −7 |
| cox3 | J | 4627–5413 | 787 | ATG | T | −1 |
| trnG | J | 5414–5474 | 61 | - | - | 0 |
| nad3 | J | 5475–5826 | 352 | ATT | T | 0 |
| trnA | J | 5828–5889 | 62 | - | - | 1 |
| trnR | J | 5895–5958 | 64 | - | - | 5 |
| trnN | J | 5959–6025 | 67 | - | - | 0 |
| trnS1 | J | 6025–6093 | 69 | - | - | −1 |
| trnE | J | 6093–6155 | 63 | - | - | −1 |
| trnF | N | 6165–6231 | 67 | - | - | 9 |
| nad5 | N | 6238–7948 | 1711 | ATG | T | 6 |
| trnH | N | 7950–8015 | 66 | - | - | 1 |
| nad4 | N | 8019–9333 | 1315 | ATG | T | 3 |
| nad4L | N | 9329–9614 | 286 | ATT | T | −5 |
| trnT | J | 9617–9679 | 63 | - | - | 2 |
| trnP | N | 9680–9742 | 63 | - | - | 0 |
| nad6 | J | 9746–10,237 | 492 | ATG | TAA | 5 |
| cytb | J | 10,237–11,368 | 1132 | ATG | T | −1 |
| trnS2 | J | 11,369–11,437 | 69 | - | - | 0 |
| nad1 | N | 11,459–12,382 | 924 | ATC | T | 21 |
| trnL1 | N | 12,383–12,449 | 67 | - | - | 0 |
| rrnL | N | 12,456–13,718 | 1263 | - | - | 6 |
| trnV | N | 13,719–13,786 | 68 | - | - | 1 |
| rrnS | N | 13,787–14,568 | 782 | - | - | 0 |
| D-−loop | J | 14,569–16,706 | 2138 | - | - | 0 |
| N. meleagris | |||||||||
| Size (bp) | T | C | A | G | A + T% | G + C% | AT Skew | GC Skew | |
| Genome | 16,199 | 30.90 | 16.40 | 42.10 | 10.60 | 73.00 | 27.00 | 0.153 | −0.215 |
| PCGs | 11,008 | 40.80 | 13.30 | 32.10 | 13.90 | 72.90 | 27.10 | −0.119 | 0.022 |
| rRNA | 2067 | 45.30 | 8.30 | 30.70 | 15.80 | 76.00 | 24.00 | −0.192 | 0.276 |
| tRNAs | 1449 | 37.40 | 10.10 | 38.80 | 13.70 | 76.20 | 23.80 | 0.018 | 0.151 |
| Control region | 1627 | 29.70 | 21.80 | 37.60 | 11.00 | 67.20 | 32.80 | 0.117 | −0.329 |
| M. harringtonae | |||||||||
| Size (bp) | T | C | A | G | A + T% | G + C% | AT skew | GC skew | |
| Genome | 15,314 | 31.80 | 15.30 | 42.60 | 10.30 | 74.50 | 25.50 | 0.145 | −0.195 |
| PCGs | 10,957 | 41.20 | 13.70 | 31.80 | 13.20 | 73.00 | 27.00 | −0.129 | −0.019 |
| rRNA | 2036 | 46.80 | 8.10 | 31.60 | 13.60 | 78.40 | 21.60 | −0.194 | 0.253 |
| tRNAs | 1439 | 39.50 | 9.30 | 37.70 | 13.50 | 77.20 | 22.80 | −0.023 | 0.184 |
| Control region | 772 | 35.50 | 12.30 | 43.80 | 8.40 | 79.30 | 20.70 | 0.105 | −0.188 |
| H. bipunctatus | |||||||||
| Size (bp) | T | C | A | G | A+T% | G+C% | AT skew | GC skew | |
| Genome | 16,706 | 32.40 | 15.00 | 41.10 | 11.60 | 73.40 | 26.60 | 0.118 | −0.128 |
| PCGs | 11,010 | 41.80 | 12.90 | 31.70 | 13.60 | 73.50 | 26.50 | −0.137 | 0.026 |
| rRNA | 2045 | 46.40 | 8.60 | 31.40 | 13.60 | 77.80 | 22.20 | −0.193 | 0.225 |
| tRNAs | 1446 | 37.30 | 10.50 | 36.80 | 15.40 | 74.10 | 25.90 | −0.007 | 0.189 |
| Control region | 2138 | 30.80 | 19.70 | 37.60 | 11.90 | 68.40 | 31.60 | 0.099 | −0.247 |



References
- Yue, Y.D.; Cao, H.Q.; Tang, F. Advance in bamboo chemical ingredients and its utilizations. J. Anhui Agric. Univ. 2007, 34, 328–333. [Google Scholar]
- Shi, J.Y.; Zhou, D.Q.; Ma, L.S.; Yao, J.; Zhang, D. Diversity of bamboo species in China. World Bamboo Rattan. 2020, 18, 55–65. [Google Scholar]
- Xiao, C.Y. Handbook for Identification of Hemiptera-Heteroptera in China Volume 1; Science Press: Beijing, China, 1977; pp. 198–268. [Google Scholar]
- Gao, C.Q.; Bu, W.J. A review of the Macropes Motschulsky (Hemiptera: Lygaeoidea: Blissidae) from China, with descriptions of three new species. Zootaxa 2010, 2366, 55–68. [Google Scholar] [CrossRef]
- Xiao, C.Y.; Ren, S.Z.; Zheng, L.Y.; Jing, X.L.; Zou, H.G.; Liu, S.L. Handbook for Identification of Hemiptera-Heteroptera in China Volume 2; Science Press: Beijing, China, 1981; pp. 43–53. [Google Scholar]
- Miyatake, T. Territorial mating aggregation in the bamboo bug, Notobitus Meleagris, Fabricius (Heteroptera: Coreidae). J. Ethol. 1995, 13, 185–189. [Google Scholar] [CrossRef]
- Miyatake, T. Multi-male mating aggregation in Notobitus meleagris (Hemiptera: Coreidae). Ann. Entomol. Soc. Am. 2002, 95, 340–344. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.Y. Studies on the biology of Notobitus meleagris. J. Appl. Entomol. 1989, 26, 226–228. [Google Scholar]
- Ding, Y.S.; Chang, Z.M.; Yang, L.; Chen, X.S. Morphological and molecular identification of Kallitaxila sinica (Walker, 1851). A New Pest of Chinese Tallow Tree. For. Res. 2018, 31, 69–75. [Google Scholar]
- Gong, N.; Yang, L.; Chen, X.S. Structural features and phylogenetic implications of four new mitogenomes of Caliscelidae (Hemiptera: Fulgoromorpha). Int. J. Mol. Sci. 2021, 22, 1348. [Google Scholar] [CrossRef]
- Zhou, Z.C.; Liu, Y.Y.; Chen, X.S. Structural features and phylogenetic implications of three new mitochondrial genomes of blister geetles (Coleoptera: Meloidae). J. Insect Sci. 2021, 21, 19. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Guo, H.; Zhu, J.; Qu, K.; Chen, Y.; Guo, Y.T.; Ding, P.; Yang, H.P.; Xu, T.; Jing, Q.; et al. Complex physical structure of complete mitochondrial genome of Quercus acutissima (Fagaceae): A significant energy plant. Genes 2022, 13, 1321. [Google Scholar] [CrossRef]
- Curole, J.P.; Kocher, T.D. Mitogenomics: Digging deeper with complete mitochondrial genomes. Trends Ecol. Evol. 1999, 14, 394–398. [Google Scholar] [CrossRef] [PubMed]
- Saccone, C.; De Giorgi, C.; Gissi, C.; Pesole, G.; Reyes, A. Evolutionary genomics in Metazoa: The mitochondrial DNA as a model system. Gene 1999, 238, 195–209. [Google Scholar] [CrossRef]
- Liu, Y.Y.; Zhou, Z.C.; Chen, X.S. Characterization of the complete mitochondrial genome of Epicauta impressicornis (Coleoptera: Meloidae) and its phylogenetic implications for the infraorder Cucujiformia. J. Insect Sci. 2020, 20, 16. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Li, H.; Song, F.; Cai, Y.; Wang, J.; Liu, J.; Cai, W. Duplication and remolding of tRNA genes in the mitochondrial genome of Reduvius tenebrosus (Hemiptera: Reduviidae). Int. J. Mol. Sci. 2016, 17, 951. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhu, R.; Xue, H.; Li, Y.; Bu, W. Mitogenomics of Chinch Bugs from China and Implications for Its Coevolutionary Relationship with Grasses. Insects 2022, 13, 643. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Wu, Y.; Liu, Y.; Zhao, P.; Chen, Z.; Song, F.; Li, H.; Cai, W. Comparative Mitogenomics and Phylogenetic Analyses of Pentatomoidea (Hemiptera: Heteroptera). Genes 2021, 12, 1306. [Google Scholar] [CrossRef] [PubMed]
- Hua, J.; Li, M.; Dong, P.; Cui, Y.; Xie, Q.; Bu, W. Comparative and phylogenomic studies on the mitochondrial genomes of Pentatomomorpha (Insecta: Hemiptera: Heteroptera). BMC Genom. 2008, 9, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, S.S.; Chen, X.S. Morphology and DNA barcode of Nisia fuliginosa Yang & Hu, 1985, A New Pest of Schoenoplectus tabernaemontani. Sichuan J. Zool. 2021, 40, 130–140. [Google Scholar]
- Xiao, C.Y. A brief introduction to the species of Cloresmini in China (Hemiptera Coreidae). Acta Entomol. Sin. 1963, 12, 124–128. [Google Scholar]
- Brailovsky, H.; Barrera, E. A revision of the Costa Rican species of Stenoeurilla Brailovsky Barrera (Hemiptera: Heteroptera: Coreidae: Stenoscelideini), with the description of two new species, new distributional records, synonymical note, and key to the known species. Zootaxa 2019, 4550, 545–556. [Google Scholar] [CrossRef]
- Tian, X.; Su, X.; Li, C.; Zhou, Y.; Li, S.; Guo, J. Draft genome of the blister beetle, Epicauta chinensis. Int. J. Biol. Macromol. 2021, 193, 1694–1706. [Google Scholar] [CrossRef] [PubMed]
- Li, H.M.; Deng, R.Q.; Wang, J.W.; Chen, Z.Y.; Jia, F.L.; Wang, X.Z. A preliminary phylogeny of the Pentatomomorpha (Hemiptera: Heteroptera) based on nuclear 18S rDNA and mitochondrial DNA sequences. Mol. Phylogenet. Evol. 2005, 37, 313–326. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, A.P.; Gao, J.; Zhao, X. Characterization of the complete mitochondrial genome of the treehopper Darthula hardwickii (Hemiptera: Aetalionidae). Mitochondrial DNA A 2016, 27, 3291. [Google Scholar] [CrossRef]
- Mu, Y.L.; Zhang, C.H.; Zhang, Y.J.; Yang, L.; Chen, X.S. Characterizing the complete mitochondrial genome of Arma custos and Picromerus lewisi (Hemiptera: Pentatomidae: Asopinae) and conducting phylogenetic analysis. J. Insect Sci. 2022, 22, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Meng, G.; Li, Y.; Yang, C.; Liu, S. MitoZ: A toolkit for animal mitochondrial genome assembly, annotation and visualization. Nucleic Acids Res. 2019, 47, e63. [Google Scholar] [CrossRef] [PubMed]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Leavengood, J.M.; Chapman, E.G.; Burkhardt, D.; Song, F.; Jiang, P. Mitochondrial phylogenomics of Hemiptera reveals adaptive innovations driving the diversification of true bugs. Proc. Biol. Sci. 2017, 284, 20171223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, T.H.; Zhang, Y.L. Two complete mitochondrial genomes of Mileewinae (Hemiptera: Cicadellidae) and a phylogenetic analysis. Insects 2021, 12, 668. [Google Scholar] [CrossRef]
- Gong, N.; Yang, L.; Chen, X.S. Comparative analysis of twelve mitogenomes of Caliscelidae (Hemiptera: Fulgoromorpha) and their phylogenetic implications. PeerJ 2021, 9, e12465. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.P.; Shan, B.B.; Liu, Y.; Wang, L.M.; Wu, Q.E.; Luo, Z.L.; Sun, D.R. Complete mitochondrial genome of two Ectoparasitic Capsalids (Platyhelminthes: Monogenea: Monopisthocotylea): Gene content, composition, and rearrangement. Genes 2022, 13, 1376. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Mena, A.; Mora, P.; Montiel, E.E.; Palomeque, T.; Lorite, P. Complete nucleotide sequence of the mitogenome of Tapinoma ibericum (Hymenoptera: Formicidae: Dolichoderinae), gene organization and phylogenetics implications for the Dolichoderinae subfamily. Genes 2022, 13, 1325. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Bivand, R.; Altman, M.; Anselin, L.; Assunção, R.; Berke, O.; Bernat, A. Spdep: Spatial Dependence: Weighting Schemes, Statistics and Models. R Package Version 1.1–2. 2019. Available online: https://cran.r-project.org/web/packages/spdep/ (accessed on 11 August 2022).
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. Muscle: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, J.; Liang, Y.Y.; Su, Y.J.; Wang, T. The complete mitochondrial genome of Ophioglossum vulgatum L. is with highly repetitive sequences: Intergenomic fragment transfer and phylogenetic analysis. Genes 2022, 13, 1287. [Google Scholar] [CrossRef]
- Vaidya, G.; Lohman, D.J.; Meier, R. Sequence matrix: Concatenation software for the fast assembly of multi-gene datasets with character set and codon information. Cladistics 2011, 27, 171–180. [Google Scholar] [CrossRef]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 2017, 34, 772–773. [Google Scholar] [CrossRef] [Green Version]
- Lanfear, R.; Calcott, B.; Ho, S.Y.; Guindon, S. PartitionFinder: Combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol. Biol. Evol. 2012, 29, 1695–1701. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.T.; Schmidt, H.A.; von, H.A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2014, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Nquyen, M.A.; von, H.A. Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 2013, 30, 1188–1195. [Google Scholar] [CrossRef] [Green Version]
- Ronquist, F.; Teslenko, M.; van der, M.P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [Green Version]
- Mousavi, S.A.; Österman, J.; Wahlberg, N.; Nesme, X.; Lavire, C.; Vial, L.; Paulin, L.; de Lajudie, P.; Lindström, K. Phylogeny of the Rhizobium-Allorhizobium-Agrobacterium clade supports the delineation of Neorhizobium gen. nov. Syst. Appl. Microbiol. 2014, 37, 208–215. [Google Scholar] [CrossRef]
- Therese, A.C.; Christopher, H.D. Molecular phylogeny of the grassland leafhopper tribe Hecalini (Hemiptera: Cicadellidae: Deltocephalinae). Ann. Entomol. Soc. Am. 2017, 111, 68–72. [Google Scholar]
- Zheng, Y.L.; Bourgoin, T.; Yang, L.; Chen, X.S.; Luo, X.Q.; Luo, G.J. Complete mitochondrial genome of the planthopper Orthopagus splendens (Germar, 1830) (Hemiptera: Fulgoromorpha: Dictyopharidae). Mitochondrial DNA B 2021, 6, 2667–2668. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yu, X.; Jiang, W.; Gao, H.; Tan, W.; Wang, W. The complete mitochondrial genome sequence of Cletus punctiger (Heteroptera: Coreidae). Mitochondrial DNA B 2019, 4, 3421–3422. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Wu, H.T.; Li, M.; Chen, W.T.; Yuan, M.L. The complete mitochondrial genome of Nysius fuscovittatus (Hemiptera: Lygaeidae). Mitochondrial DNA B 2020, 5, 3483–3484. [Google Scholar] [CrossRef] [PubMed]
- Sureshan, S.C.; Tanavade, R.V.; Ghosh, S.; Ghosh, S.; Sella, R.N.; Mohideen, H.S. Complete mitochondrial genome sequencing of Oxycarenus laetus (Hemiptera: Lygaeidae) from two geographically distinct regions of India. Sci. Rep. 2021, 11, 23738. [Google Scholar] [CrossRef]
- Forthman, M.; Miller, C.W.; Kimball, R.T. Phylogenomic analysis with improved taxon sampling corroborates an Alydidae+Hydarinae+Pseudophloeinae clade (Heteroptera: Coreoidea: Alydidae, Coreidae). Org. Divers. Evol. 2022, 22, 669–679. [Google Scholar] [CrossRef]
- Song, N.; Liang, A.P.; Bu, C.P. A molecular phylogeny of Hemiptera inferred from mitochondrial genome sequences. PLoS ONE 2012, 7, e48778. [Google Scholar] [CrossRef] [Green Version]
- Yao, Y.Z.; Ren, D. Phylogeny, origin and evolution of Pentatomomorpha. Abstract Volume. In Proceedings of the 10th National Congress of Palaeontological Society of China (PSC)-The 25th Annual Conference of PSC, Nanjing, China, 13–17 October 2009. [Google Scholar]
- Zhu, G.L.; Wang, Q.R.; Zheng, Z.M. Phylogenetic relationship among ten species of Riptortus linearis in Coreinae (Hemiptera: Coreidae) based on EST Isozyme. J. Anhui Agric. Sci. 2011, 39, 11462–11463, 11466. [Google Scholar]
- Zhang, Q.L.; Feng, R.Q.; Li, M.; Guo, Z.L.; Zhang, L.J.; Luo, F.Z.; Cao, Y.; Yuan, M.L. The complete mitogenome of Pyrrhocoris tibialis (Hemiptera: Pyrrhocoridae) and phylogenetic implications. Genes 2019, 10, 820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, X.; Wang, K.B.; Tang, Z.C.; Zhang, Y.; Yi, W.; Xue, H. Phylogeny of Coreoidea based on mitochondrial genomes show the paraphyly of Coreidae and Alydidae. Arch. Insect Biochem. Physiol. 2022, 110, e21878. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.M. Fauna Editorial Committee Academia Sinica. Economic Insect Fauna of China Fasc. 31 Hemiptera (1); Science Press: Beijing, China, 1985; pp. 117–159. [Google Scholar]
- Xu, G.Y.; Yang, A.N.; Yang, S.D.; Zhang, Y.H.; Chen, C.S. Studies on biology characteristic observation and control experiment in Pirkimerus japonicus Hidaka. J. Anhui Agric. Sci. 2007, 35, 4884–4958. [Google Scholar]
- Gao, Z.W. Studies on the morphology, life cycle and control methods of Pirkimerus japonicus. J. Zhejiang For. Sci. Technol. 1980, 000, 5–7. [Google Scholar]
- Haldhar, S.M. Report of Homoeocerus variabilis (Hemiptera: Coreidae) on khejri (Prosopis cineraria) in Rajasthan, India: Incidence and Morphometric Analysis. Fla. Entomol. Int. J. Am. 2012, 95, 848–853. [Google Scholar] [CrossRef]
- Long, Z.Q. Bionomics and occurrence of Notobius Meleagris in Guizhou. Chin. Bull. Entomol. 2009, 46, 133–135. [Google Scholar]
- Wang, Y.; Huang, X.L.; Qiao, G.X. Comparative analysis of mitochondrial genomes of five aphid species (Hemiptera: Aphididae) and phylogenetic implications. PLoS ONE 2013, 8, e77511. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Yang, J.; Li, Y.W.; Cui, Y.; Xie, Q.; Bu, W.J.; Hillis, D.M. A mitochondrial genome of Rhyparochromidae (Hemiptera: Heteroptera) and a comparative analysis of related mitochondrial genomes. Sci. Rep. 2016, 6, 35175. [Google Scholar] [CrossRef] [Green Version]
- Wang, J. Comparative Mitogenomics and Genetic Diversity of Mirid Bugs (Hemiptera: Miridae). Master’s Thesis, Lanzhou University, Lanzhou, China, 2017. [Google Scholar]
- Zhao, L.; Wei, J.F.; Zhao, W.Q.; Chen, C.; Gao, X.Y.; Zhao, Q. The complete mitochondrial genome of Pentatoma rufipes (Hemiptera, Pentatomidae) and its phylogenetic implications. ZooKeys 2021, 1042, 51–72. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, H.X.; Yu, X.F.; Yang, M.F. Comparative analysis of mitochondrial genomes among twelve sibling species of the genus Atkinsoniella Distant, 1908 (Hemiptera: Cicadellidae: Cicadellinae) and phylogenetic analysis. Insects 2022, 13, 254. [Google Scholar] [CrossRef]
- Song, F.; Li, H.; Shao, R.F.; Shi, A.M.; Bai, X.S.; Zheng, X.R.; Heiss, E.; Cai, W.Z. Rearrangement of mitochondrial tRNA genes in flat bugs (Hemiptera: Aradidae). Sci. Rep. 2016, 6, 25725. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.L.; Chen, Q.D.; Chen, S.; Pu, D.Q.; Chen, Z.T.; Liu, Y.Y.; Liu, X. The highly rearranged mitochondrial genomes of three economically important scale insects and the mitochondrial phylogeny of Coccoidea (Hemiptera: Sternorrhyncha). PeerJ 2020, 8, e9932. [Google Scholar] [CrossRef]
- Ye, F.; Li, H.; Xie, Q. Mitochondrial genomes from two specialized subfamilies of Reduviidae (Insecta: Hemiptera) reveal novel gene rearrangements of true bugs. Genes 2021, 12, 1134. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, H.Y.; Cao, L.G.; Shi, A.M.; Yang, H.L.; Cai, W.Z. The complete mitochondrial genome of the damsel bug Alloeorhynchus bakeri (Hemiptera: Nabidae). Int. J. Biol. Sci. 2012, 8, 93–107. [Google Scholar] [CrossRef] [Green Version]
- Li, H.U.; Liu, H.; Song, F.; Shi, A.; Zhou, X.; Cai, W. Comparative mitogenomic analysis of damsel bugs representing three tribes in the family Nabidae (Insecta: Hemiptera). PLoS ONE 2012, 7, 1–9. [Google Scholar] [CrossRef]
- Schmidt, J.M.; Barney, S.K.; Williams, M.A.; Bessin, R.T.; Coolong, T.W.; Harwood, J.D. Predator–prey trophic relationships in response to organic management practices. Mol. Ecol. 2014, 23, 3777–3789. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, W.; Yang, L.; Long, J.; Chang, Z.; Gong, N.; Mu, Y.; Lv, S.; Chen, X. Characterizing the Complete Mitochondrial Genomes of Three Bugs (Hemiptera: Heteroptera) Harming Bamboo. Genes 2023, 14, 342. https://doi.org/10.3390/genes14020342
Zhu W, Yang L, Long J, Chang Z, Gong N, Mu Y, Lv S, Chen X. Characterizing the Complete Mitochondrial Genomes of Three Bugs (Hemiptera: Heteroptera) Harming Bamboo. Genes. 2023; 14(2):342. https://doi.org/10.3390/genes14020342
Chicago/Turabian StyleZhu, Wenli, Lin Yang, Jiankun Long, Zhimin Chang, Nian Gong, Yinlin Mu, Shasha Lv, and Xiangsheng Chen. 2023. "Characterizing the Complete Mitochondrial Genomes of Three Bugs (Hemiptera: Heteroptera) Harming Bamboo" Genes 14, no. 2: 342. https://doi.org/10.3390/genes14020342