
SERPINB3, Adult-Onset Immunodeficiency, and Generalized Pustular Psoriasis
 , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
2.1.1. Patient 1
2.1.2. Patient 2
2.1.3. Patient 3
2.2. Whole-Exome Sequencing and Mutation Analysis
2.3. Histopathology
2.4. Immunohistochemistry
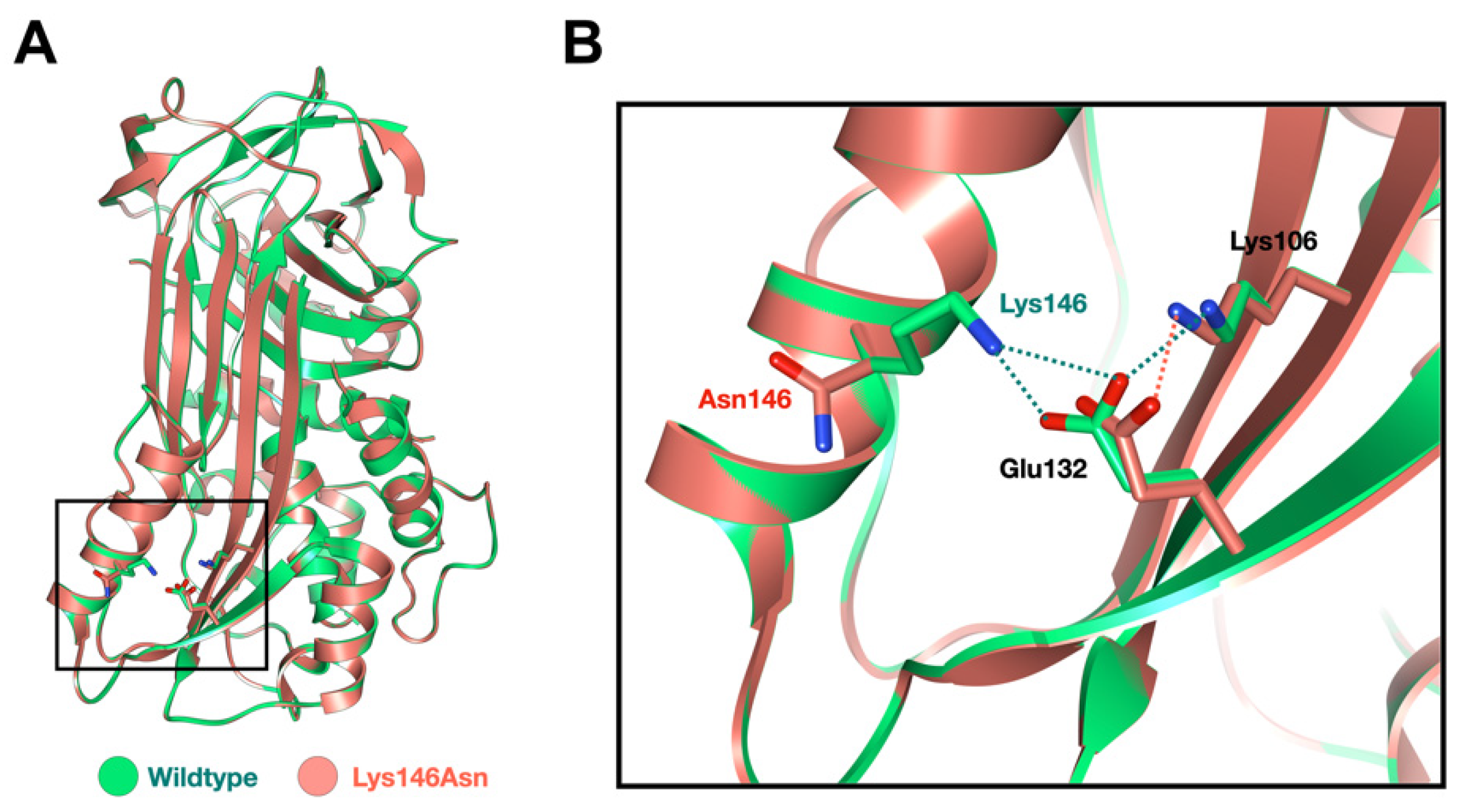
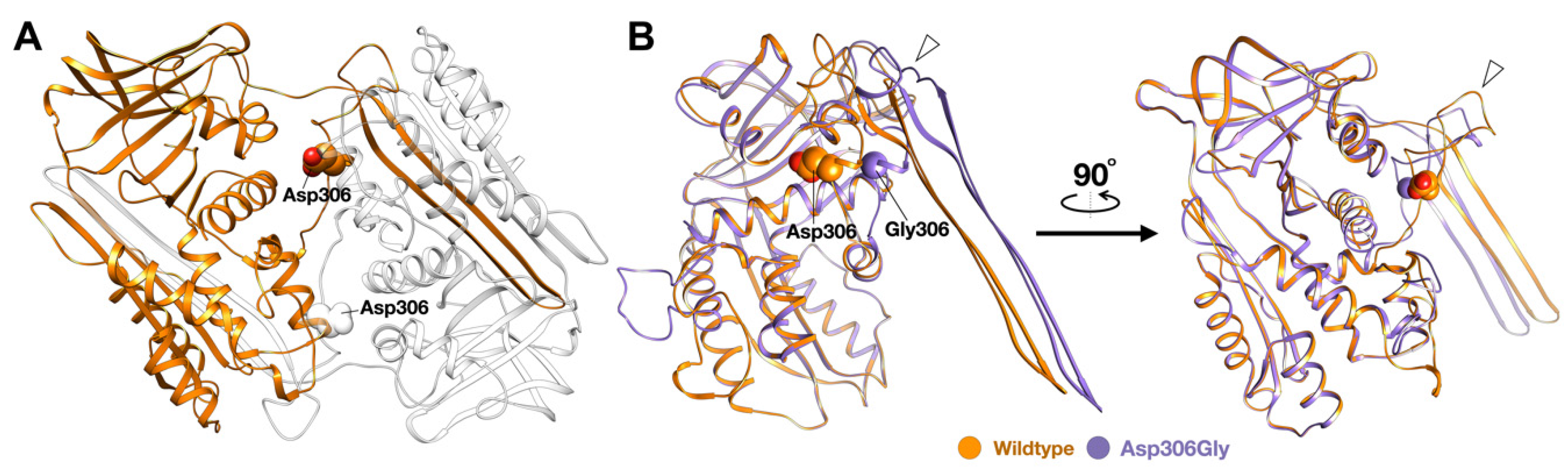
2.5. Molecular Modeling of the SERPINB3 Variants
3. Results
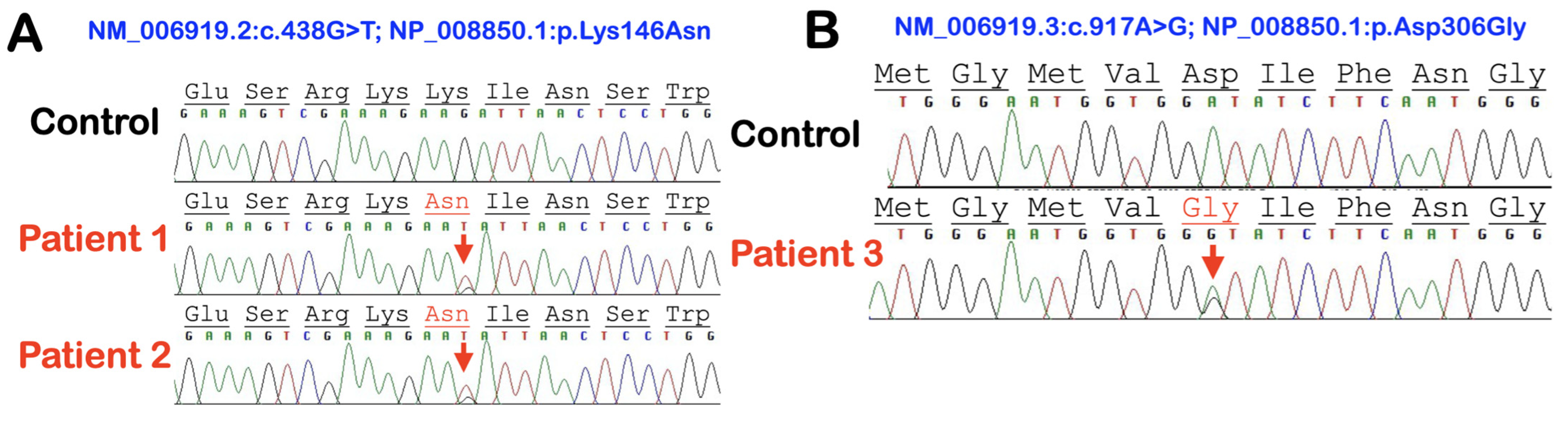
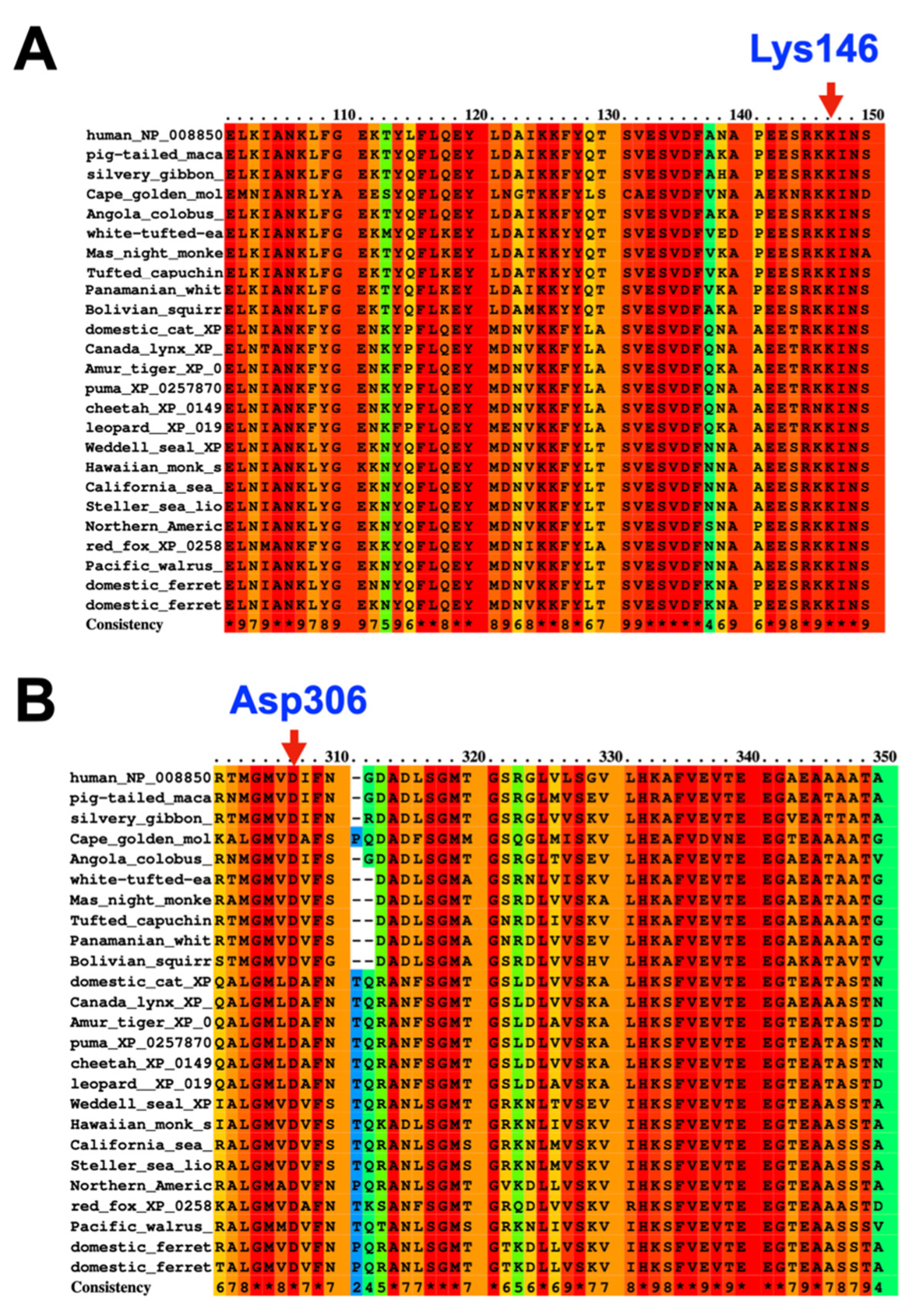
3.1. Whole-Exome Sequence Sequencing and Bioinformatic Analysis
3.2. Histopathological Findings
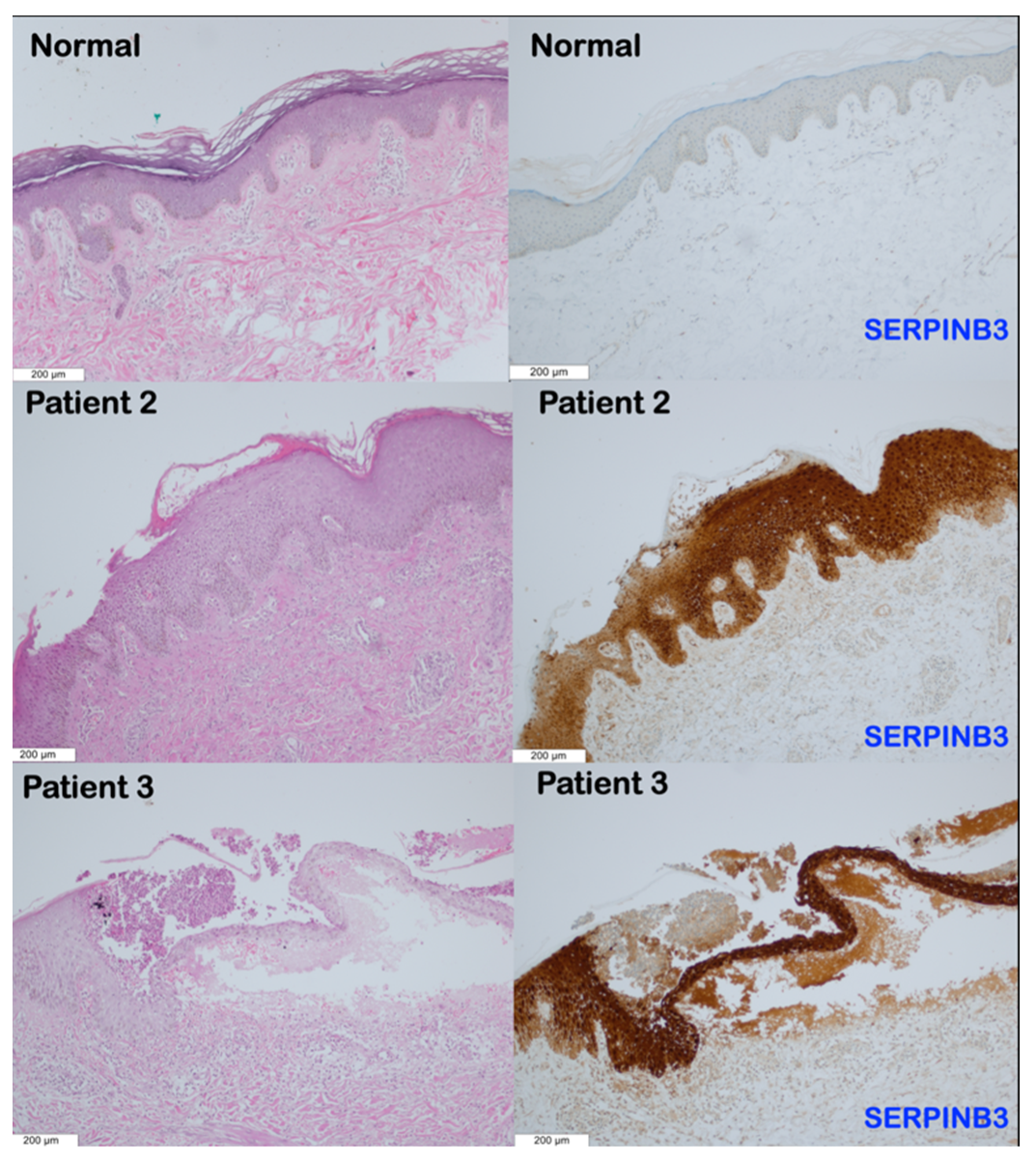
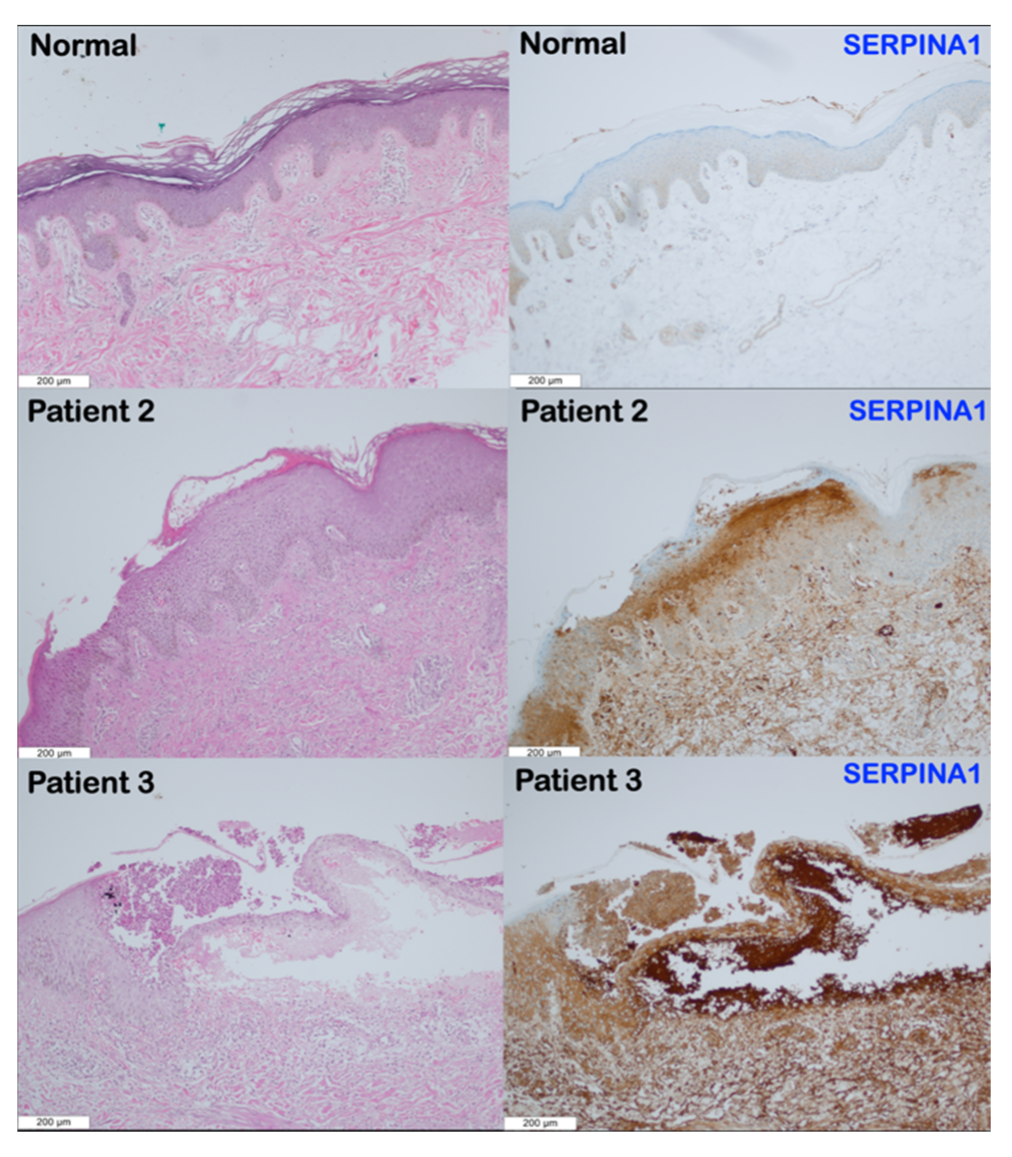
3.3. Immunohistochemical Findings
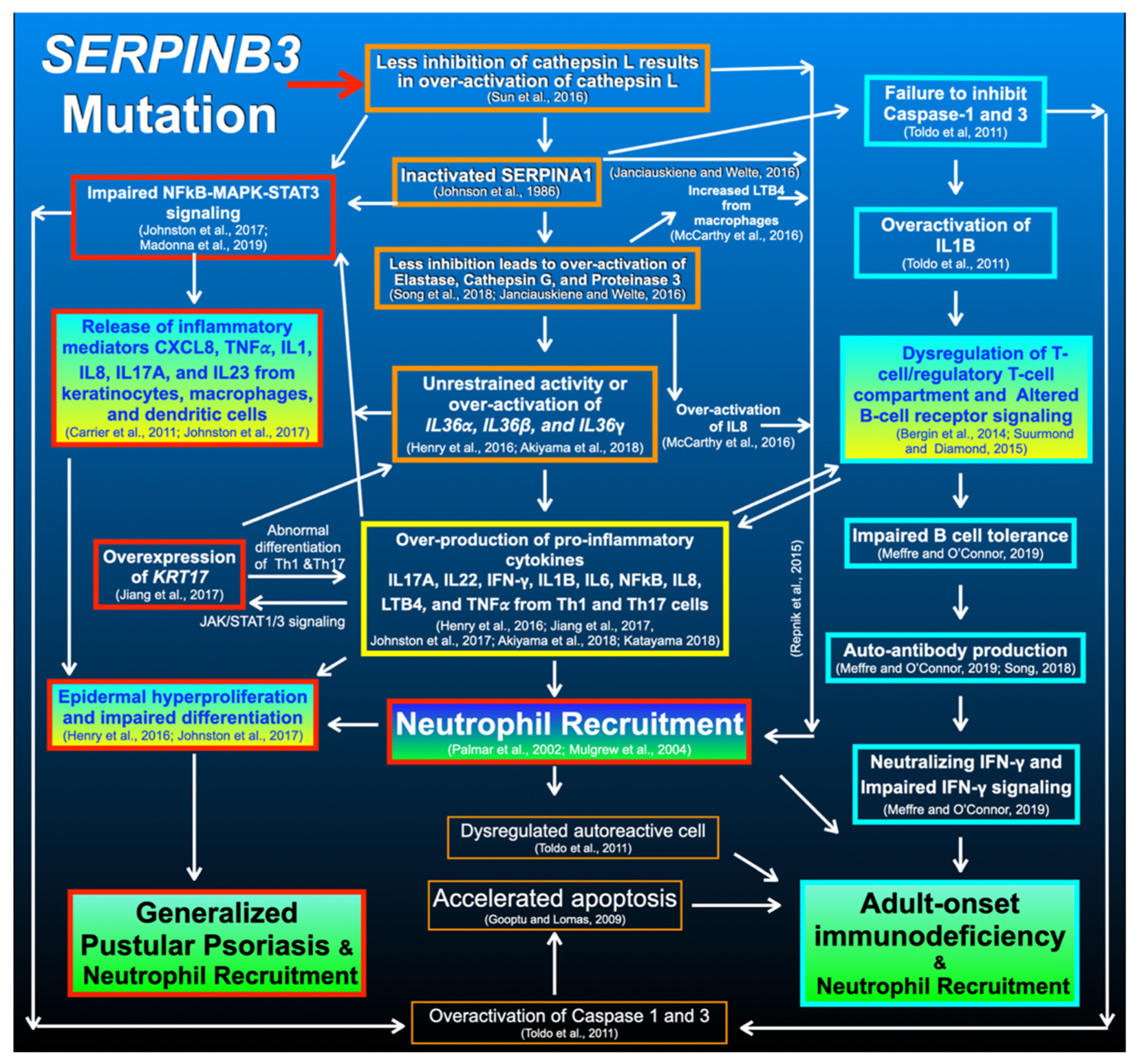
4. Discussion
4.1. SERPINB3 Variants and GPP and AOID Phenotypes
4.2. SERPINB3 Variant Predisposed Patients to GPP
4.3. SERPINB3 Variants Predispose Patients to AOID
4.4. Overexpression of SERPINA1 and SERPINB3 in Patients with SERPINB3 Variants
4.5. Overproduction of IFN-γ Autoantibodies in Patients with AOID
4.6. SERPINB3 Mutations, Pso p27, and Pustular Skin Reaction
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jutivorakool, K.; Sittiwattanawong, P.; Kantikosum, K.; Hurst, C.P.; Kumtornrut, C.; Asawanonda, P.; Klaewsongkram, J.; Rerknimitr, P. Skin Manifestations in Patients with Adult-onset Immunodeficiency due to Anti-interferon-gamma Autoantibody: A Relationship with Systemic Infections. Acta Dermatol. Venereol. 2018, 98, 742–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uppala, R.; Tsoi, L.C.; Harms, P.W.; Wang, B.; Billi, A.C.; Maverakis, E.; Michelle Kahlenberg, J.; Ward, N.L.; Gudjonsson, J.E. “Autoinflammatory psoriasis”-genetics and biology of pustular psoriasis. Cell. Mol. Immunol. 2021, 18, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Kantaputra, P.; Chaowattanapanit, S.; Kiratikanon, S.; Chaiwarith, R.; Choonhakarn, C.; Intachai, W.; Quarto, N.; Tongsima, S.; Ketudat Cairns, J.R.; Ngamphiw, C.; et al. SERPINA1, generalized pustular psoriasis, and adult-onset immunodeficiency. J. Dermatol. 2021, 48, 1597–1601. [Google Scholar] [CrossRef] [PubMed]
- Frey, S.; Sticht, H.; Wilsmann-Theis, D.; Gerschütz, A.; Wolf, K.; Löhr, S.; Haskamp, S.; Frey, B.; Hahn, M.; Ekici, A.B.; et al. Rare Loss-of-Function Mutation in SERPINA3 in Generalized Pustular Psoriasis. J. Investig. Dermatol. 2020, 140, 1451–1455.e1413. [Google Scholar] [CrossRef] [PubMed]
- Kantaputra, P.N.; Chuamanochan, M.; Kiratikanon, S.; Chiewchanvit, S.; Chaiwarith, R.; Intachai, W.; Quarto, N.; Tongsima, S.; McGrath, J.A.; Ngamphiw, C. A truncating variant in SERPINA3, skin pustules and adult-onset immunodeficiency. J. Dermatol. 2021, 48, e370–e371. [Google Scholar] [CrossRef]
- Kaiserman, D.; Whisstock, J.C.; Bird, P.I. Mechanisms of serpin dysfunction in disease. Expert Rev. Mol. Med. 2006, 8, 1–19. [Google Scholar] [CrossRef]
- Mangan, M.S.; Kaiserman, D.; Bird, P.I. The role of serpins in vertebrate immunity. Tissue Antigens 2008, 72, 1–10. [Google Scholar] [CrossRef]
- Correnti, M.; Cappon, A.; Pastore, M.; Piombanti, B.; Lori, G.; Oliveira, D.; Munoz-Garrido, P.; Lewinska, M.; Andersen, J.B.; Coulouarn, C.; et al. The protease-inhibitor SerpinB3 as a critical modulator of the stem-like subset in human cholangiocarcinoma. Liver Int. 2022, 42, 233–248. [Google Scholar] [CrossRef]
- Kato, H. Expression and function of squamous cell carcinoma antigen. Anticancer Res. 1996, 16, 2149–2153. [Google Scholar]
- Suminami, Y.; Nagashima, S.; Vujanovic, N.L.; Hirabayashi, K.; Kato, H.; Whiteside, T.L. Inhibition of apoptosis in human tumour cells by the tumour-associated serpin, SCC antigen-1. Br. J. Cancer 2000, 82, 981–989. [Google Scholar] [CrossRef] [Green Version]
- Takeda, A.; Higuchi, D.; Takahashi, T.; Ogo, M.; Baciu, P.; Goetinck, P.F.; Hibino, T. Overexpression of serpin squamous cell carcinoma antigens in psoriatic skin. J. Investig. Dermatol. 2002, 118, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Gatto, M.; Luisetto, R.; Ghirardello, A.; Cavicchioli, L.; Codolo, G.; Biasiolo, A.; Maggioni, G.; Saccon, F.; Beggio, M.; Cappon, A.; et al. SERPINB3 Delays Glomerulonephritis and Attenuates the Lupus-Like Disease in Lupus Murine Models by Inducing a More Tolerogenic Immune Phenotype. Front. Immunol. 2018, 9, 2081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iversen, O.J.; Lysvand, H.; Slupphaug, G. Pso p27, a SERPINB3/B4-derived protein, is most likely a common autoantigen in chronic inflammatory diseases. Clin. Immunol. 2017, 174, 10–17. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [Green Version]
- Lysvand, H.; Helland, R.; Hagen, L.; Slupphaug, G.; Iversen, O.J. Psoriasis pathogenesis-Pso p27 constitutes a compact structure forming large aggregates. Biochem. Biophys. Rep. 2015, 2, 132–136. [Google Scholar] [CrossRef] [Green Version]
- Yamasaki, M.; Li, W.; Johnson, D.J.; Huntington, J.A. Crystal structure of a stable dimer reveals the molecular basis of serpin polymerization. Nature 2008, 455, 1255–1258. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–w303. [Google Scholar] [CrossRef] [Green Version]
- Williams, C.J.; Headd, J.J.; Moriarty, N.W.; Prisant, M.G.; Videau, L.L.; Deis, L.N.; Verma, V.; Keedy, D.A.; Hintze, B.J.; Chen, V.B.; et al. MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 2018, 27, 293–315. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.F.; Kaushik, A.; Kapil, C.; Gupta, D.; Jairajpuri, M.A. A hydrophobic patch surrounding Trp154 in human neuroserpin controls the helix F dynamics with implications in inhibition and aggregation. Sci. Rep. 2017, 7, 42987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkar, A.; Zhou, C.; Meklemburg, R.; Wintrode, P.L. Local conformational flexibility provides a basis for facile polymer formation in human neuroserpin. Biophys. J. 2011, 101, 1758–1765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabrita, L.D.; Whisstock, J.C.; Bottomley, S.P. Probing the role of the F-helix in serpin stability through a single tryptophan substitution. Biochemistry 2002, 41, 4575–4581. [Google Scholar] [CrossRef]
- Sun, Y.; Sheshadri, N.; Zong, W.X. SERPINB3 and B4: From biochemistry to biology. Semin. Cell Dev. Biol. 2017, 62, 170–177. [Google Scholar] [CrossRef] [Green Version]
- Schick, C.; Pemberton, P.A.; Shi, G.P.; Kamachi, Y.; Cataltepe, S.; Bartuski, A.J.; Gornstein, E.R.; Brömme, D.; Chapman, H.A.; Silverman, G.A. Cross-class inhibition of the cysteine proteinases cathepsins K, L, and S by the serpin squamous cell carcinoma antigen 1: A kinetic analysis. Biochemistry 1998, 37, 5258–5266. [Google Scholar] [CrossRef]
- Sivaprasad, U.; Askew, D.J.; Ericksen, M.B.; Gibson, A.M.; Stier, M.T.; Brandt, E.B.; Bass, S.A.; Daines, M.O.; Chakir, J.; Stringer, K.F.; et al. A nonredundant role for mouse Serpinb3a in the induction of mucus production in asthma. J. Allergy Clin. Immunol. 2011, 127, 254–261. [Google Scholar] [CrossRef] [Green Version]
- Johnson, D.A.; Barrett, A.J.; Mason, R.W. Cathepsin L inactivates alpha 1-proteinase inhibitor by cleavage in the reactive site region. J. Biol. Chem. 1986, 261, 14748–14751. [Google Scholar] [CrossRef]
- Johnston, A.; Xing, X.; Wolterink, L.; Barnes, D.H.; Yin, Z.; Reingold, L.; Kahlenberg, J.M.; Harms, P.W.; Gudjonsson, J.E. IL-1 and IL-36 are dominant cytokines in generalized pustular psoriasis. J. Allergy Clin. Immunol. 2017, 140, 109–120. [Google Scholar] [CrossRef] [Green Version]
- Akiyama, M.; Takeichi, T.; McGrath, J.A.; Sugiura, K. Autoinflammatory keratinization diseases: An emerging concept encompassing various inflammatory keratinization disorders of the skin. J. Dermatol. Sci. 2018, 90, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Henry, C.M.; Sullivan, G.P.; Clancy, D.M.; Afonina, I.S.; Kulms, D.; Martin, S.J. Neutrophil-Derived Proteases Escalate Inflammation through Activation of IL-36 Family Cytokines. Cell Rep. 2016, 14, 708–722. [Google Scholar] [CrossRef] [PubMed]
- Song, S. Alpha-1 Antitrypsin Therapy for Autoimmune Disorders. Chronic Obs. Pulm. Dis. 2018, 5, 289–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, M.; Sun, Z.; Dang, E.; Li, B.; Fang, H.; Li, J.; Gao, L.; Zhang, K.; Wang, G. TGFβ/SMAD/microRNA-486-3p Signaling Axis Mediates Keratin 17 Expression and Keratinocyte Hyperproliferation in Psoriasis. J. Investig. Dermatol. 2017, 137, 2177–2186. [Google Scholar] [CrossRef] [Green Version]
- Janciauskiene, S.; Welte, T. Well-Known and Less Well-Known Functions of Alpha-1 Antitrypsin. Its Role in Chronic Obstructive Pulmonary Disease and Other Disease Developments. Ann. Am. Thorac. Soc. 2016, 13 (Suppl. 4), S280–S288. [Google Scholar] [CrossRef]
- Vidalino, L.; Doria, A.; Quarta, S.; Zen, M.; Gatta, A.; Pontisso, P. SERPINB3, apoptosis and autoimmunity. Autoimmun. Rev. 2009, 9, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Bergin, D.A.; Reeves, E.P.; Hurley, K.; Wolfe, R.; Jameel, R.; Fitzgerald, S.; McElvaney, N.G. The circulating proteinase inhibitor α-1 antitrypsin regulates neutrophil degranulation and autoimmunity. Sci. Transl. Med. 2014, 6, 217ra211. [Google Scholar] [CrossRef] [PubMed]
- Meffre, E.; O’Connor, K.C. Impaired B-cell tolerance checkpoints promote the development of autoimmune diseases and pathogenic autoantibodies. Immunol. Rev. 2019, 292, 90–101. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, C.; Reeves, E.P.; McElvaney, N.G. The Role of Neutrophils in Alpha-1 Antitrypsin Deficiency. Ann. Am. Thorac. Soc. 2016, 13 (Suppl. 4), S297–S304. [Google Scholar] [CrossRef]
- Parmar, J.S.; Mahadeva, R.; Reed, B.J.; Farahi, N.; Cadwallader, K.A.; Keogan, M.T.; Bilton, D.; Chilvers, E.R.; Lomas, D.A. Polymers of alpha(1)-antitrypsin are chemotactic for human neutrophils: A new paradigm for the pathogenesis of emphysema. Am. J. Respir. Cell Mol. Biol. 2002, 26, 723–730. [Google Scholar] [CrossRef]
- Mulgrew, A.T.; Taggart, C.C.; Lawless, M.W.; Greene, C.M.; Brantly, M.L.; O’Neill, S.J.; McElvaney, N.G. Z alpha1-antitrypsin polymerizes in the lung and acts as a neutrophil chemoattractant. Chest 2004, 125, 1952–1957. [Google Scholar] [CrossRef]
- Suurmond, J.; Diamond, B. Autoantibodies in systemic autoimmune diseases: Specificity and pathogenicity. J. Clin. Investig. 2015, 125, 2194–2202. [Google Scholar] [CrossRef]
- Katayama, H. Development of psoriasis by continuous neutrophil infiltration into the epidermis. Exp. Dermatol. 2018, 27, 1084–1091. [Google Scholar] [CrossRef] [Green Version]
- Gooptu, B.; Lomas, D.A. Conformational pathology of the serpins: Themes, variations, and therapeutic strategies. Annu. Rev. Biochem. 2009, 78, 147–176. [Google Scholar] [CrossRef]
- Toldo, S.; Seropian, I.M.; Mezzaroma, E.; Van Tassell, B.W.; Salloum, F.N.; Lewis, E.C.; Voelkel, N.; Dinarello, C.A.; Abbate, A. Alpha-1 antitrypsin inhibits caspase-1 and protects from acute myocardial ischemia-reperfusion injury. J. Mol. Cell. Cardiol. 2011, 51, 244–251. [Google Scholar] [CrossRef]
- Madonna, S.; Girolomoni, G.; Dinarello, C.A.; Albanesi, C. The Significance of IL-36 Hyperactivation and IL-36R Targeting in Psoriasis. Int. J. Mol. Sci. 2019, 20, 3318. [Google Scholar] [CrossRef] [Green Version]
- Carrier, Y.; Ma, H.L.; Ramon, H.E.; Napierata, L.; Small, C.; O′Toole, M.; Young, D.A.; Fouser, L.A.; Nickerson-Nutter, C.; Collins, M.; et al. Inter-regulation of Th17 cytokines and the IL-36 cytokines in vitro and in vivo: Implications in psoriasis pathogenesis. J. Investig. Dermatol. 2011, 131, 2428–2437. [Google Scholar] [CrossRef] [Green Version]
- Repnik, U.; Starr, A.E.; Overall, C.M.; Turk, B. Cysteine Cathepsins Activate ELR Chemokines and Inactivate Non-ELR Chemokines. J. Biol. Chem. 2015, 290, 13800–13811. [Google Scholar] [CrossRef] [Green Version]
- Gatto, M.; Iaccarino, L.; Ghirardello, A.; Bassi, N.; Pontisso, P.; Punzi, L.; Shoenfeld, Y.; Doria, A. Serpins, immunity and autoimmunity: Old molecules, new functions. Clin. Rev. Allergy Immunol. 2013, 45, 267–280. [Google Scholar] [CrossRef]
- Vidalino, L.; Doria, A.; Quarta, S.M.; Crescenzi, M.; Ruvoletto, M.; Frezzato, F.; Trentin, L.; Turato, C.; Parolin, M.C.; Ghirardello, A.; et al. SERPINB3 expression on B-cell surface in autoimmune diseases and hepatitis C virus-related chronic liver infection. Exp. Biol. Med. 2012, 237, 793–802. [Google Scholar] [CrossRef]
- Jakoš, T.; Pišlar, A.; Jewett, A.; Kos, J. Cysteine Cathepsins in Tumor-Associated Immune Cells. Front. Immunol. 2019, 10, 2037. [Google Scholar] [CrossRef] [Green Version]
- Badano, M.N.; Camicia, G.L.; Lombardi, G.; Maglioco, A.; Cabrera, G.; Costa, H.; Meiss, R.P.; Piazzon, I.; Nepomnaschy, I. B-cell lymphopoiesis is regulated by cathepsin L. PLoS ONE 2013, 8, e61347. [Google Scholar] [CrossRef]
- Katagiri, C.; Nakanishi, J.; Kadoya, K.; Hibino, T. Serpin squamous cell carcinoma antigen inhibits UV-induced apoptosis via suppression of c-JUN NH2-terminal kinase. J. Cell Biol. 2006, 172, 983–990. [Google Scholar] [CrossRef] [Green Version]
- Luke, C.J.; Pak, S.C.; Askew, Y.S.; Naviglia, T.L.; Askew, D.J.; Nobar, S.M.; Vetica, A.C.; Long, O.S.; Watkins, S.C.; Stolz, D.B.; et al. An intracellular serpin regulates necrosis by inhibiting the induction and sequelae of lysosomal injury. Cell 2007, 130, 1108–1119. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, K.; Kiyoshima, T.; Matsuo, K.; Ozeki, S.; Sakai, H. Effect of SCCA1 and SCCA2 on the suppression of TNF-alpha-induced cell death by impeding the release of mitochondrial cytochrome c in an oral squamous cell carcinoma cell line. Tumour Biol. 2005, 26, 165–172. [Google Scholar] [CrossRef]
- Turato, C.; Pontisso, P. SERPINB3 (serpin peptidase inhibitor, clade B (ovalbumin), member 3). Atlas Genet. Cytogenet. Oncol. Haematol. 2015, 19, 202–209. [Google Scholar] [CrossRef]
- Turato, C.; Biasiolo, A.; Pengo, P.; Frecer, V.; Quarta, S.; Fasolato, S.; Ruvoletto, M.; Beneduce, L.; Zuin, J.; Fassina, G.; et al. Increased antiprotease activity of the SERPINB3 polymorphic variant SCCA-PD. Exp. Biol. Med. 2011, 236, 281–290. [Google Scholar] [CrossRef]
- Iversen, O.J.; Bergh, K.; Lysvand, H. Use of scale antibodies for the detection of antigens in psoriatic lesions. Acta Dermatol. Venereol. 1993, 73, 31–34. [Google Scholar]
- Asbakk, K.; Bergh, K.; Iversen, O.J. The psoriasis-associated antigen, pso p27, participates in the formation of complement activating immune-complexes in psoriatic scale. Apmis 1990, 98, 143–149. [Google Scholar] [CrossRef]
- Paterson, M.A.; Horvath, A.J.; Pike, R.N.; Coughlin, P.B. Molecular characterization of centerin, a germinal centre cell serpin. Biochem. J. 2007, 405, 489–494. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patients | Dx | SERPINB3 Variants | SERPINA5 Variant | SERPINA9 Variant | IL1RN Variant | IL1RL2 Variant |
|---|---|---|---|---|---|---|
| 1 | GPP | chr18: g.61325778C>A NM_006919.2: c.438G>T NP_008850.1 p.Lys146Asn rs193238900 Allele freq = 0.0002126 | ||||
| 2 | AOID | chr14:g.94929551G>A NM_001042518.2 c.833C>T NP_001035983.2 p.Ser278Leu rs370040760 Allele freq = 0.00006406 | NM_000577.5 c.392C>T NP_000568.1 p.Ala131Val rs374276440 Allele freq = 0.00003183 | NM_003854.2 c.791G>A NP_003845.2 p.Arg264Lys Allele freq = 0.0001875 | ||
| 3 | AOID | chr18: g.61323147T>C NM_006919.2 c.917A>G NP_008850.1 p.Asp306Gly NOVEL | NM_000624.5 c.175G>A NP_000615.3 p.Ala59Thr rs139961453 Allele freq = 0.0003658 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kantaputra, P.; Daroontum, T.; Chuamanochan, M.; Chaowattanapanit, S.; Kiratikanon, S.; Choonhakarn, C.; Intachai, W.; Olsen, B.; Tongsima, S.; Ngamphiw, C.; et al. SERPINB3, Adult-Onset Immunodeficiency, and Generalized Pustular Psoriasis. Genes 2023, 14, 266. https://doi.org/10.3390/genes14020266
Kantaputra P, Daroontum T, Chuamanochan M, Chaowattanapanit S, Kiratikanon S, Choonhakarn C, Intachai W, Olsen B, Tongsima S, Ngamphiw C, et al. SERPINB3, Adult-Onset Immunodeficiency, and Generalized Pustular Psoriasis. Genes. 2023; 14(2):266. https://doi.org/10.3390/genes14020266
Chicago/Turabian StyleKantaputra, Piranit, Teerada Daroontum, Mati Chuamanochan, Suteeraporn Chaowattanapanit, Salin Kiratikanon, Charoen Choonhakarn, Worrachet Intachai, Bjorn Olsen, Sissades Tongsima, Chumpol Ngamphiw, and et al. 2023. "SERPINB3, Adult-Onset Immunodeficiency, and Generalized Pustular Psoriasis" Genes 14, no. 2: 266. https://doi.org/10.3390/genes14020266