
Spectrum of Causative Mutations in Patients with Hemophilia A in Russia
 ,
, 
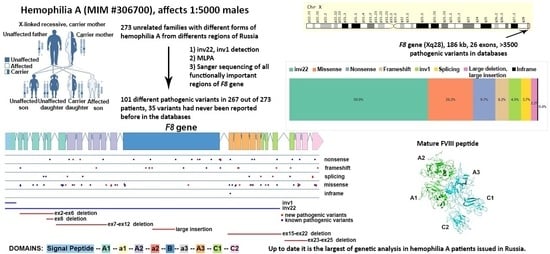
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Patient Samples
2.2. DNA Collection and Extraction
2.3. Detection of Mutations
2.3.1. PCR Detection of Intron 1 and 22 Inversion
2.3.2. Amplification of the F8 Gene
2.3.3. Large Deletions/Insertions Detection
2.3.4. Pathogenic Variant Evaluation
2.3.5. Variant Nomenclature
3. Results
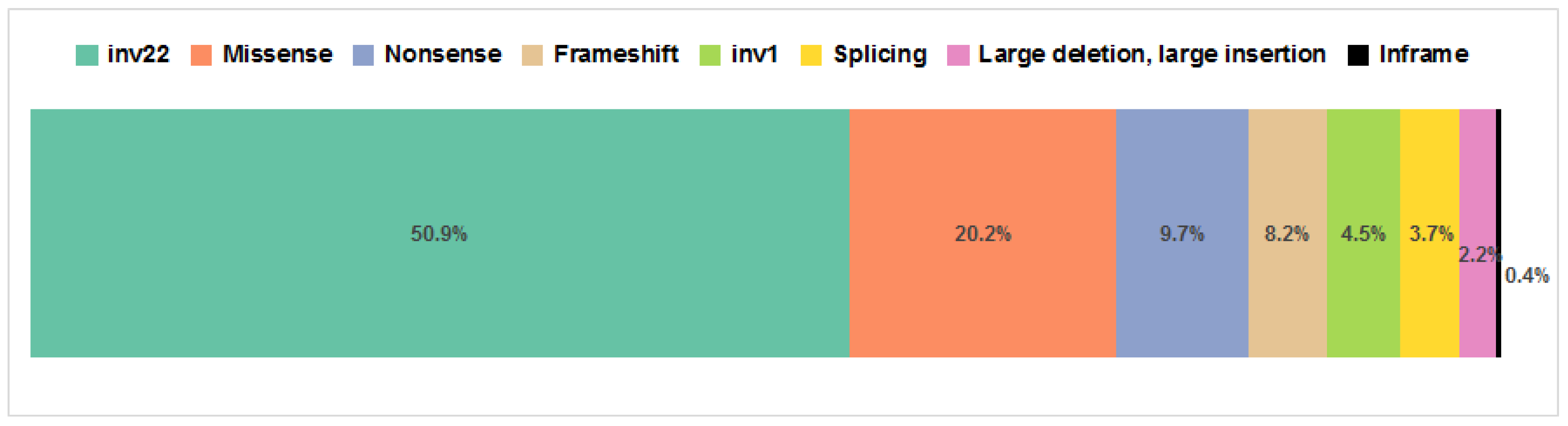
3.1. Inversions
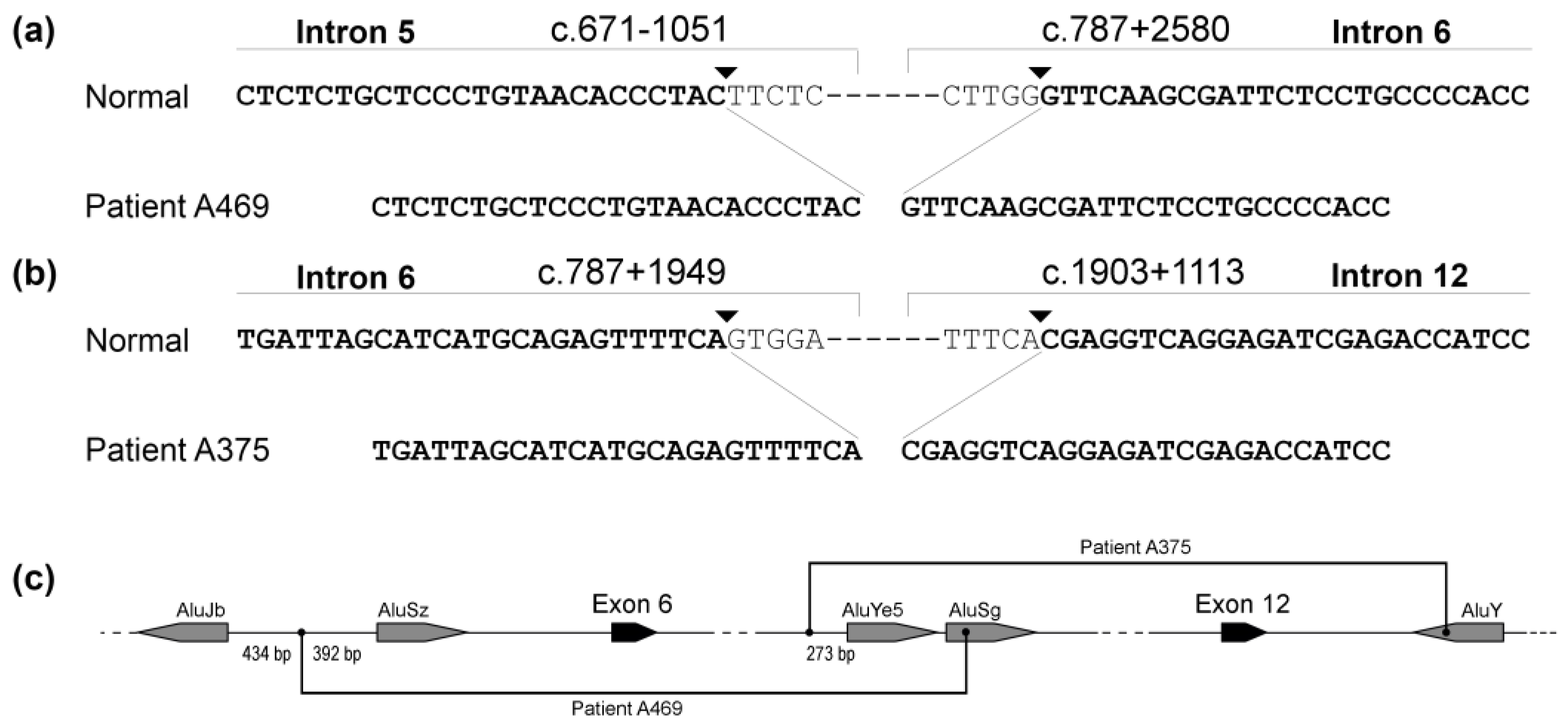
3.2. Large Deletions and Insertions
3.3. Loss-of-Function Variants
3.4. Splicing Variants
3.5. Missense Mutations and Inframe Deletion
3.6. Previously Undescribed Variants Assessment
4. Discussion
4.1. Clinical Manifestations of Studied Patients
4.2. Patients without Genetic Variants in F8 Gene
4.3. Patients with Two Genetic Variants
4.4. Localization of Point Variants
4.5. Large Deletions
4.6. De Novo Origin Evaluation
4.7. Inhibitor Development
- High risk of inhibitor development: large deletions (several exons), nonsense mutations in the light chain (A3C1C2 domains);
- Moderate risk: large deletions (one exon), nonsense mutations in the heavy chain (A1A2B domains), inv22, inv1;
- Low risk: microdeletions/microinsertions, missense mutations, splicing mutations [63].
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gitschier, J.; Wood, W.I.; Goralka, T.M.; Wion, K.L.; Chen, E.Y.; Eaton, D.H.; Vehar, G.A.; Capon, D.J.; Lawn, R.M. Characterization of the Human Factor VIII Gene. Nature 1984, 312, 326–330. [Google Scholar] [CrossRef]
- Fang, H.; Wang, L.; Wang, H. The Protein Structure and Effect of Factor VIII. Thromb. Res. 2007, 119, 1–13. [Google Scholar] [CrossRef]
- Astermark, J. FVIII Inhibitors: Pathogenesis and Avoidance. Blood 2015, 125, 2045–2051. [Google Scholar] [CrossRef] [Green Version]
- Astermark, J.; Donfield, S.M.; Gomperts, E.D.; Schwarz, J.; Menius, E.D.; Pavlova, A.; Oldenburg, J.; Kessing, B.; DiMichele, D.M.; Shapiro, A.D.; et al. The Polygenic Nature of Inhibitors in Hemophilia A: Results from the Hemophilia Inhibitor Genetics Study (HIGS) Combined Cohort. Blood 2013, 121, 1446–1454. [Google Scholar] [CrossRef] [Green Version]
- Bardi, E.; Astermark, J. Genetic Risk Factors for Inhibitors in Haemophilia A. Eur. J. Haematol. 2015, 94, 7–10. [Google Scholar] [CrossRef] [Green Version]
- Minno, G.D. Eptacog Alfa Activated: A Recombinant Product to Treat Rare Congenital Bleeding Disorders. Blood Rev. 2015, 29, S26–S33. [Google Scholar] [CrossRef]
- Lakich, D.; Kazazian, H.H.; Antonarakis, S.E.; Gitschier, J. Inversions Disrupting the Factor VIII Gene Are a Common Cause of Severe Haemophilia A. Nat. Genet. 1993, 5, 236–241. [Google Scholar] [CrossRef]
- Bagnall, R.D.; Waseem, N.; Green, P.M.; Giannelli, F. Recurrent Inversion Breaking Intron 1 of the Factor VIII Gene Is a Frequent Cause of Severe Hemophilia A. Blood 2002, 99, 168–174. [Google Scholar] [CrossRef] [Green Version]
- Atik, T.; Işık, E.; Onay, H.; Akgün, B.; Kavaklı, K.; Evim, M.; Tuysuz, G.; Ozbek, N.Y.; Şahin, F.; Salcıoğlu, Z.; et al. Factor 8 Gene Mutation Spectrum of 270 Patients with Haemophilia A: Identification of 36 Novel Mutations. Turk. J. Hematol. 2020, 37, 145–153. [Google Scholar] [CrossRef]
- Abdul-Ghafar, A.; Bogdanova, N.; Lim, L.C.; Zhao, Y.; Markoff, A.; Tien, S.L. Ten Novel Factor VIII (F8C) Mutations in Eighteen Haemophilia A Families Detected in Singapor. Haemophilia 2009, 16, 551–553. [Google Scholar] [CrossRef]
- Albánez, S.; Ruiz-Sáez, A.; Boadas, A.; De Bosch, N.; Porco, A. Identification of Factor VIII Gene Mutations in Patients with Severe Haemophilia A in Venezuela: Identification of Seven Novel Mutations. Haemophilia 2011, 17, e913–e918. [Google Scholar] [CrossRef]
- Awidi, A.; Ramahi, M.; Al Hattab, D.; Mefleh, R.; Dweiri, M.; Bsoul, N.; Magablah, A.; Arafat, E.; Barqawi, M.; Bishtawi, M.; et al. Study of Mutations in Jordanian Patients with Haemophilia A: Identification of Five Novel Mutations. Haemophilia 2010, 16, 136–142. [Google Scholar] [CrossRef]
- Azadmehr, S.; Rahiminejad, F.; Zafarghandi Motlagh, F.; Jamali, M.; Ghazizadeh Tehrani, P.; Shirzadeh, T.; Bagherian, H.; Karimipoor, M.; Davoudi-Dehaghani, E.; Zeinali, S. The Spectrum of Pathogenic Variants in Iranian Families with Hemophilia A. Arch. Iran. Med. 2021, 24, 887–896. [Google Scholar] [CrossRef]
- Borràs, N.; Castillo-González, D.; Comes, N.; Martin-Fernandez, L.; Rivero-Jiménez, R.A.; Chang-Monteagudo, A.; Ruiz-Moleón, V.; Garrote-Santana, H.; Vidal, F.; Macías-Abraham, C. Molecular Study of a Large Cohort of 109 Haemophilia Patients from Cuba Using a Gene Panel with next Generation Sequencing-based Technology. Haemophilia 2022, 28, 125–137. [Google Scholar] [CrossRef]
- Feng, Y.; Li, Q.; Shi, P.; Liu, N.; Kong, X.; Guo, R. Mutation Analysis in the F8 Gene in 485 Families with Haemophilia A and Prenatal Diagnosis in China. Haemophilia 2021, 27, e88–e92. [Google Scholar] [CrossRef]
- Green, P.M.; Bagnall, R.D.; Waseem, N.H.; Giannelli, F. Haemophilia A Mutations in the UK: Results of Screening One-Third of the Population. Br. J. Haematol. 2008, 143, 115–128. [Google Scholar] [CrossRef]
- Nair, P.S.; Shetty, S.; Kulkarni, B.; Ghosh, K. Molecular Pathology of Haemophilia A in Indian Patients: Identification of 11 Novel Mutations. Clin. Chim. Acta 2010, 411, 2004–2008. [Google Scholar] [CrossRef]
- Owaidah, T.M.; Alkhail, H.A.; Zahrani, H.A.; Musa, A.A.; Saleh, M.A.; Riash, M.A.; Alodaib, A.; Amero, K.A. Molecular Genotyping of Hemophilia A in Saudi Arabia: Report of 2 Novel Mutations. Blood Coagul. Fibrinolysis 2009, 20, 415–418. [Google Scholar] [CrossRef]
- Pinto, P.; Ghosh, K.; Shetty, S. F8 Gene Mutation Profile in Indian Hemophilia A Patients: Identification of 23 Novel Mutations and Factor VIII Inhibitor Risk Association. Mutat. Res. Mol. Mech. Mutagen. 2016, 786, 27–33. [Google Scholar] [CrossRef]
- Reitter, S.; Sturn, R.; Horvath, B.; Freitag, R.; Male, C.; Muntean, W.; Streif, W.; Pabinger, I.; Mannhalter, C.; Austrian Molecular Haemophilia Study Group. Spectrum of Causative Mutations in Patients with Haemophilia A in Austria. Thromb. Haemost. 2010, 104, 78–85. [Google Scholar] [CrossRef]
- Riccardi, F.; Tagliaferri, A.; Martorana, D.; Rivolta, G.F.; Valdrè, L.; Rodorigo, G.; Biasoli, C.; D’Incà, M.; Serino, M.L.; Macchi, S.; et al. Spectrum of F8 Gene Mutations in Haemophilia A Patients from a Region of Italy: Identification of 23 New Mutations. Haemophilia 2010, 16, 791–800. [Google Scholar] [CrossRef] [PubMed]
- Santacroce, R.; Acquila, M.; Belvini, D.; Castaldo, G.; Garagiola, I.; Giacomelli, S.H.; Lombardi, A.M.; Minuti, B.; Riccardi, F.; Salviato, R.; et al. Identification of 217 Unreported Mutations in the F8 Gene in a Group of 1,410 Unselected Italian Patients with Hemophilia A. J. Hum. Genet. 2008, 53, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Santacroce, R.; Santoro, R.; Sessa, F.; Iannaccaro, P.; Sarno, M.; Longo, V.; Gallone, A.; Vecchione, G.; Muleo, G.; Margaglione, M. Screening of Mutations of Hemophilia A in 40 Italian Patients: A Novel G-to-A Mutation in Intron 10 of the F8 Gene as a Putative Cause of Mild Hemophilia a in Southern Italy. Blood Coagul. Fibrinolysis 2008, 19, 197–202. [Google Scholar] [CrossRef]
- Venceslá, A.; Corral-Rodríguez, M.Á.; Baena, M.; Cornet, M.; Domènech, M.; Baiget, M.; Fuentes-Prior, P.; Tizzano, E.F. Identification of 31 Novel Mutations in the F8 Gene in Spanish Hemophilia A Patients: Structural Analysis of 20 Missense Mutations Suggests New Intermolecular Binding Sites. Blood 2008, 111, 3468–3478. [Google Scholar] [CrossRef]
- Villarreal-Martínez, L.; Ibarra-Ramirez, M.; Calvo-Anguiano, G.; Lugo-Trampe, J.D.J.; Luna-Záizar, H.; Martínez-de-Villarreal, L.E.; Meléndez-Aranda, L.; Jaloma-Cruz, A.-R. Molecular Genetic Diagnosis by Next-Generation Sequencing in a Cohort of Mexican Patients with Haemophilia and Report of Novel Variants. Blood Cells. Mol. Dis. 2020, 83, 102423. [Google Scholar] [CrossRef]
- Xue, F.; Zhang, L.; Sui, T.; Ge, J.; Gu, D.; Du, W.; Zhao, H.; Yang, R. Factor VIII Gene Mutations Profile in 148 Chinese Hemophilia A Subjects: Factor VIII Gene Mutations Profile. Eur. J. Haematol. 2010, 85, 264–272. [Google Scholar] [CrossRef]
- Beskorovainaya, T.S.; Milovidova, T.B.; Schagina, O.A.; Ryzhkova, O.P.; Polyakov, A.V. Complex Molecular Diagnostics of Hemophilia A in Russian Patients. Russ. J. Genet. 2019, 55, 1015–1024. [Google Scholar] [CrossRef]
- White, G.C.; Rosendaal, F.; Aledort, L.M.; Lusher, J.M.; Rothschild, C.; Ingerslev, J. Recommendation of the Scientific Subcommittee on Factor VIII and Factor IX of the Scientific and Standardization Committee of the International Society on Thrombosis and Haemostasis. Thromb. Haemost. 2001, 85, 560. [Google Scholar]
- Maniatis, T.; Fritsch, E.F.; Sambrook, J. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory: Cold Spring Harbor, NY, USA, 1982; ISBN 978-0-87969-136-3. [Google Scholar]
- Liu, Q.; Nozari, G.; Sommer, S.S. Single-Tube Polymerase Chain Reaction for Rapid Diagnosis of the Inversion Hotspot of Mutation in Hemophilia A. Blood 1998, 92, 1458–1459. [Google Scholar] [CrossRef]
- Bagnall, R.D.; Giannelli, F.; Green, P.M. Int22h-Related Inversions Causing Hemophilia A: A Novel Insight into Their Origin and a New More Discriminant PCR Test for Their Detection. J. Thromb. Haemost. 2006, 4, 591–598. [Google Scholar] [CrossRef]
- Salomashkina, V.V.; Pshenichnikova, O.S.; Perina, F.G.; Surin, V.L. A Founder Effect in Hemophilia A Patients from Russian Ural Region with a New p.(His634Arg) Variant in F8 Gene. Blood Coagul. Fibrinolysis 2022, 33, 124–129. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A Method and Server for Predicting Damaging Missense Mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, Y.; Sims, G.E.; Murphy, S.; Miller, J.R.; Chan, A.P. Predicting the Functional Effect of Amino Acid Substitutions and Indels. PLoS ONE 2012, 7, e46688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sim, N.-L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT Web Server: Predicting Effects of Amino Acid Substitutions on Proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation Prediction for the Deep-Sequencing Age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef]
- Rentzsch, P.; Schubach, M.; Shendure, J.; Kircher, M. CADD-Splice—Improving Genome-Wide Variant Effect Prediction Using Deep Learning-Derived Splice Scores. Genome Med. 2021, 13, 31. [Google Scholar] [CrossRef]
- Hamasaki-Katagiri, N.; Salari, R.; Wu, A.; Qi, Y.; Schiller, T.; Filiberto, A.C.; Schisterman, E.F.; Komar, A.A.; Przytycka, T.M.; Kimchi-Sarfaty, C. A Gene-Specific Method for Predicting Hemophilia-Causing Point Mutations. J. Mol. Biol. 2013, 425, 4023–4033. [Google Scholar] [CrossRef] [Green Version]
- Laskowski, R.A.; Stephenson, J.D.; Sillitoe, I.; Orengo, C.A.; Thornton, J.M. VarSite: Disease Variants and Protein Structure. Protein Sci. 2020, 29, 111–119. [Google Scholar] [CrossRef] [Green Version]
- Ittisoponpisan, S.; Islam, S.A.; Khanna, T.; Alhuzimi, E.; David, A.; Sternberg, M.J.E. Can Predicted Protein 3D Structures Provide Reliable Insights into Whether Missense Variants Are Disease Associated? J. Mol. Biol. 2019, 431, 2197–2212. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Chernetskaya, D.M.; Surin, V.L.; Salomashkina, V.V.; Pshenichnikova, O.S.; Yakovleva, E.V.; Zozulya, N.I.; Sudarikov, A.B.; Likhacheva, E.A.; Shabanova, E.S.; Perina, F.G. Molecular and Genetic Verification of von Willebrand Disease Type 2N. Russ. J. Hematol. Transfusiology 2022, 67, 172–180. [Google Scholar] [CrossRef]
- Beskorovainaya, T.S.; Milovidova, T.B.; Shchagina, O.A.; Matushchenko, E.G.; Petuhova, M.S.; Togochakova, O.K.; Salomashkina, V.V.; Pshenichnikova, O.S.; Surin, V.L.; Polyakov, A.V.; et al. Complex F8 Gene Rearrangements. Mol. Med. 2019, 17, 41–51. [Google Scholar] [CrossRef]
- David, D.; Ventura, C.; Moreira, I.; Diniz, M.J.; Antunes, M.; Tavares, A.; Araújo, F.; Morais, S.; Campos, M.; Lavinha, J.; et al. The Spectrum of Mutations and Molecular Pathogenesis of Hemophilia A in 181 Portuguese Patients. Haematologica 2006, 91, 840–843. [Google Scholar]
- Bogdanova, N.; Markoff, A.; Pollmann, H.; Nowak-Göttl, U.; Eisert, R.; Dworniczak, B.; Eigel, A.; Horst, J. Prevalence of Small Rearrangements in the Factor VIII Gene F8C among Patients with Severe Hemophilia A. Hum. Mutat. 2002, 20, 236–237. [Google Scholar] [CrossRef] [PubMed]
- Fernández-López, O.; García-Lozano, J.-R.; Núñez-Vázquez, R.; Pérez-Garrido, R.; Núñez-Roldán, A. The Spectrum of Mutations in Southern Spanish Patients with Hemophilia A and Identification of 28 Novel Mutations. Haematologica 2005, 90, 707–710. [Google Scholar]
- Guillet, B.; Lambert, T.; d’Oiron, R.; Proulle, V.; Plantier, J.-L.; Rafowicz, A.; Peynet, J.; Costa, J.-M.; Bendelac, L.; Laurian, Y.; et al. Detection of 95 Novel Mutations in Coagulation Factor VIII Gene F8 Responsible for Hemophilia A: Results from a Single Institution. Hum. Mutat. 2006, 27, 676–685. [Google Scholar] [CrossRef]
- Johnsen, J.M.; Fletcher, S.N.; Huston, H.; Roberge, S.; Martin, B.K.; Kircher, M.; Josephson, N.C.; Shendure, J.; Ruuska, S.; Koerper, M.A.; et al. Novel Approach to Genetic Analysis and Results in 3000 Hemophilia Patients Enrolled in the My Life, Our Future Initiative. Blood Adv. 2017, 1, 824–834. [Google Scholar] [CrossRef] [Green Version]
- Klopp, N.; Oldenburg, J.; Uen, C.; Schneppenheim, R.; Graw, J. 11 Hemophilia A Patients without Mutations in the Factor VIII Encoding Gene. Thromb. Haemost. 2002, 88, 357–360. [Google Scholar] [CrossRef]
- Vinciguerra, C.; Zawadzki, C.; Dargaud, Y.; Pernod, G.; Berger, C.; Nougier, C.; Négrier, C. Characterisation of 96 Mutations in 128 Unrelated Severe Haemophilia A Patients from France: Description of 62 Novel Mutations. Thromb. Haemost. 2006, 95, 593–599. [Google Scholar] [CrossRef] [Green Version]
- Bach, E.; Wolf, B.; Oldenburg, J.; Müller, C.; Rost, S. Identification of Deep Intronic Variants in 15 Haemophilia A Patients by next Generation Sequencing of the Whole Factor VIII Gene. Thromb. Haemost. 2015, 114, 757–767. [Google Scholar] [CrossRef] [Green Version]
- Bagnall, R.D.; Waseem, N.H.; Green, P.M.; Colvin, B.; Lee, C.; Giannelli, F. Creation of a Novel Donor Splice Site in Intron 1 of the Factor VIII Gene Leads to Activation of a 191 Bp Cryptic Exon in Two Haemophilia A Patients. Br. J. Haematol. 1999, 107, 766–771. [Google Scholar] [CrossRef] [PubMed]
- Castaman, G.; Giacomelli, S.H.; Mancuso, M.E.; D’Andrea, G.; Santacroce, R.; Sanna, S.; Santagostino, E.; Mannucci, P.M.; Goodeve, A.; Rodeghiero, F. Deep Intronic Variations May Cause Mild Hemophilia A: Intronic Mutations in Mild Hemophilia A. J. Thromb. Haemost. 2011, 9, 1541–1548. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.; Perng, C.; Cheng, S.; Hu, S.; Wu, T.; Lin, S.; Chen, Y. Deep Intronic Variant c.5999-277G>A of F8 Gene May Be a Hot Spot Mutation for Mild Hemophilia A Patients without Mutation in Exonic DNA. Eur. J. Haematol. 2019, 103, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Inaba, H.; Koyama, T.; Shinozawa, K.; Amano, K.; Fukutake, K. Identification and Characterization of an Adenine to Guanine Transition within Intron 10 of the Factor VIII Gene as a Causative Mutation in a Patient with Mild Haemophilia A. Haemophilia 2013, 19, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Jourdy, Y.; Janin, A.; Fretigny, M.; Lienhart, A.; Négrier, C.; Bozon, D.; Vinciguerra, C. Reccurrent F8 Intronic Deletion Found in Mild Hemophilia A Causes Alu Exonization. Am. J. Hum. Genet. 2018, 102, 199–206. [Google Scholar] [CrossRef] [Green Version]
- Pezeshkpoor, B.; Zimmer, N.; Marquardt, N.; Nanda, I.; Haaf, T.; Budde, U.; Oldenburg, J.; El-Maarri, O. Deep Intronic ‘Mutations’ Cause Hemophilia A: Application of next Generation Sequencing in Patients without Detectable Mutation in F8 CDNA. J. Thromb. Haemost. 2013, 11, 1679–1687. [Google Scholar] [CrossRef] [Green Version]
- El-Maarri, O.; Herbiniaux, U.; Graw, J.; Schroder, J.; Terzic, A.; Watzka, M.; Brackmann, H.H.; Schramm, W.; Hanfland, P.; Schwaab, R.; et al. Analysis of MRNA in Hemophilia A Patients with Undetectable Mutations Reveals Normal Splicing in the Factor VIII Gene. J. Thromb. Haemost. 2005, 3, 332–339. [Google Scholar] [CrossRef]
- Jankowska, K.I.; McGill, J.; Pezeshkpoor, B.; Oldenburg, J.; Atreya, C.D.; Sauna, Z.E. Clinical Manifestation of Hemophilia A in the Absence of Mutations in the F8 Gene That Encodes FVIII: Role of MicroRNAs. Transfusion 2020, 60, 401–413. [Google Scholar] [CrossRef]
- Jourdy, Y.; Chatron, N.; Fretigny, M.; Dericquebourg, A.; Sanlaville, D.; Vinciguerra, C. Comprehensive Analysis of F8 Large Deletions: Characterization of Full Breakpoint Junctions and Description of a Possible DNA Breakage Hotspot in Intron 6. J. Thromb. Haemost. 2022, 20, 2293–2305. [Google Scholar] [CrossRef]
- Gouw, S.C.; van den Berg, H.M.; Oldenburg, J.; Astermark, J.; de Groot, P.G.; Margaglione, M.; Thompson, A.R.; van Heerde, W.; Boekhorst, J.; Miller, C.H.; et al. F8 Gene Mutation Type and Inhibitor Development in Patients with Severe Hemophilia A: Systematic Review and Meta-Analysis. Blood 2012, 119, 2922–2934. [Google Scholar] [CrossRef] [Green Version]
- Schwaab, R.; Pavlova, A.; Albert, T.; Caspers, M.; Oldenburg, J. Significance of F8 Missense Mutations with Respect to Inhibitor Formation. Thromb. Haemost. 2013, 109, 464–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garagiola, I.; Palla, R.; Peyvandi, F. Risk factors for inhibitor development in severe hemophilia A. Thromb. Haemost. 2018, 168, 20–27. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Severe/Moderate | Mild | ND 1 | Total | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Inh | No Inh | ND | Total | Inh | No Inh | ND | Total | ND | Total | ||
| Inv22 | 20 | 23 | 79 | 122 | 2 | 2 | 12 | 12 | 136 | ||
| Inv1 | 1 | 6 | 4 | 11 | 1 | 1 | 0 | 12 | |||
| Large deletions | 4 | 1 | 5 | 0 | 0 | 5 | |||||
| Large insertions | 1 | 1 | 0 | 0 | 1 | ||||||
| Frameshift | 2 | 2 | 12 | 16 | 3 | 3 | 3 | 3 | 22 | ||
| Nonsense | 7 | 7 | 9 | 23 | 0 | 3 | 3 | 26 | |||
| Splicing | 1 | 1 | 7 | 9 | 0 | 1 | 1 | 10 | |||
| Missense | 1 | 15 | 13 | 29 | 1 | 9 | 11 | 21 | 4 | 4 | 54 |
| Inframe | 1 | 1 | 0 | 0 | 1 | ||||||
| Pathogenic Variant | Nucleotide Change Type | AA Effect | N Patients | Pathogenicity [41] |
|---|---|---|---|---|
| c.926C>T p.(Pro309Leu) | SNV | M | 1 | Uncertain significance (PM2, PP2, PP3) |
| c.1007A>G p.(His336Arg) | SNV | M | 1 | Uncertain significance (PM2, PP2, PP3, PP4, BP2) |
| c.1010-2A>G | SNV | S | 1 | Pathogenic (PVS1, PM2, PP3, PP4) |
| c.1195A>T p.(Lys399*) | SNV | N | 1 | Pathogenic (PVS1, PM2, PP4) |
| c.1366A>T p.(Lys456*) | SNV | N | 1 | Pathogenic (PVS1, PM2, PP4) |
| c.1633delC p.(Pro545Leufs*4) | Deletion | F | 1 | Likely pathogenic (PVS1, PM2) |
| c.1911T>A p.(Asn637Lys) | SNV | M | 1 | Likely pathogenic (PM1, PM2, PM5, PP2, PP3, PP4) |
| c.1921T>C p.(Phe641Leu) | SNV | M | 1 | Likely pathogenic (PM2, PM5, PP2, PP3) |
| c.2156G>T p.(Arg719Ile) | SNV | M | 1 | Pathogenic (PS2, PM2, PM5, PP2, PP3, PP4) |
| c.2830delA p.(Lys943Serfs*12) | Deletion | F | 1 | Pathogenic (PVS1, PM2, PP4) |
| c.3031_3032del p.(Lys1011Serfs*1) | Deletion | F | 1 | Likely pathogenic (PVS1, PM2) |
| c.3794C>T p.(Pro1265Leu) | SNV | M | 1 | Uncertain significance (PM2, PP2, PP4, BP2, BP4) |
| c.4089_4090delinsTT p.(Met1363Ile*) | Indel | N | 1 | Pathogenic (PVS1, PM2, PP4) |
| c.4834A>T p.(Lys1612*) | SNV | N | 1 | Likely pathogenic (PVS1, PM2) |
| c.4836_4837delinsAT p.(Lys1613*) | Indel | N | 1 | Pathogenic (PVS1, PM2, PP4) |
| c.5413T>C p.(Tyr1805His) | SNV | M | 1 | Likely pathogenic (PM2, PM5, PP2, PP3) |
| c.5475dupT p.(Val1826Cysfs*4) | Insertion | F | 1 | Likely pathogenic (PVS1, PM2) |
| c.5509delinsTGCAACATCATATGGCAACATCATAT p.(Lys1837Cysfs*17) | Complex indel | F | 1 | Pathogenic (PVS1, PS2, PM2) |
| c.5533A>G p.(Thr1845Ala) | SNV | M | 2 | Uncertain significance (PM2, PM5, PP2, BP4) |
| c.5878C>G p.(Arg1960Gly) | SNV | M | 1 | Likely pathogenic (PM1, PM2, PM5, PP2, PP3) |
| c.6193T>A p.(Trp2065Arg) | SNV | M | 1 | Pathogenic (PS1, PM2, PM5, PP2, PP3) |
| c.6274-2A>G | SNV | S | 1 | Pathogenic (PVS1, PM2, PP3, PP4) |
| c.6285_6291delinsTAATTGGTAGGTG p.(Leu2095Phefs*3) | Complex indel | F | 1 | Pathogenic (PVS1, PM2, PP4) |
| c.6442delA p.(Asn2148Metfs*7) | Deletion | F | 1 | Pathogenic (PVS1, PM2, PP4) |
| c.6634T>G p.(Ser2212Ala) | SNV | M | 1 | Likely pathogenic (PM2, PM5, PP2, PP3) |
| c.6664T>C p.(Trp2222Arg) | SNV | M | 1 | Likely pathogenic (PS1, PM2, PP2, PP3, PP4) |
| c.7013T>C p.(Leu2338Pro) | SNV | M | 1 | Uncertain significance (PM2, PP2, PP3, PP4) |
| c.7021G>C p.(Glu2341Gln) | SNV | M | 1 | Likely pathogenic (PM2, PM5, PP2, PP3, PP4) |
| c.7041_7068delinsC p.(Asp2349Hisfs*29) | Complex indel | F | 1 | Likely pathogenic (PVS1, PM2) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pshenichnikova, O.; Salomashkina, V.; Poznyakova, J.; Selivanova, D.; Chernetskaya, D.; Yakovleva, E.; Dimitrieva, O.; Likhacheva, E.; Perina, F.; Zozulya, N.; et al. Spectrum of Causative Mutations in Patients with Hemophilia A in Russia. Genes 2023, 14, 260. https://doi.org/10.3390/genes14020260
Pshenichnikova O, Salomashkina V, Poznyakova J, Selivanova D, Chernetskaya D, Yakovleva E, Dimitrieva O, Likhacheva E, Perina F, Zozulya N, et al. Spectrum of Causative Mutations in Patients with Hemophilia A in Russia. Genes. 2023; 14(2):260. https://doi.org/10.3390/genes14020260
Chicago/Turabian StylePshenichnikova, Olesya, Valentina Salomashkina, Julia Poznyakova, Daria Selivanova, Daria Chernetskaya, Elena Yakovleva, Oksana Dimitrieva, Elena Likhacheva, Farida Perina, Nadezhda Zozulya, and et al. 2023. "Spectrum of Causative Mutations in Patients with Hemophilia A in Russia" Genes 14, no. 2: 260. https://doi.org/10.3390/genes14020260