
Framework of the Alu Subfamily Evolution in the Platyrrhine Three-Family Clade of Cebidae, Callithrichidae, and Aotidae


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. History of Alu Elements
1.2. Alu Analyses of Whole Genomes
1.3. Alu Evolution in Platyrrhini
2. Materials and Methods
2.1. Lineage-Specific Alu Elements
2.2. Alu Subfamily Analysis
2.3. Model Selection
2.4. SCULU Analysis
2.5. Bayesian Phylogenetic Analysis
3. Results
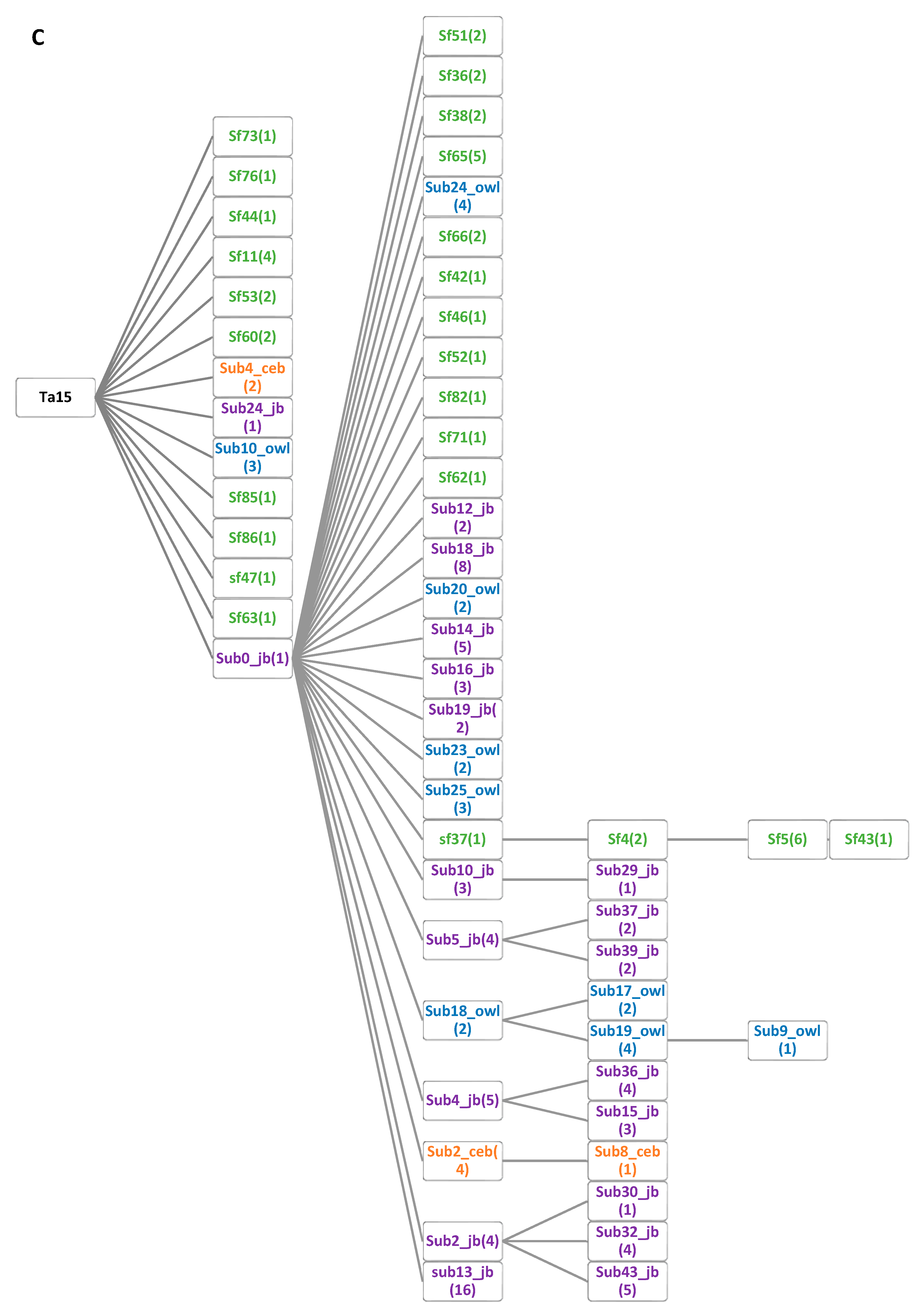
3.1. Owl Monkey and Capuchin Alu Element Subfamilies
3.2. Alu Subfamily Network Analysis
3.3. Alu Subfamily Phylogenetic Analysis
3.4. Computationally Distinct Subfamilies
3.5. Number of Lineage Specific Alu Elements by Subfamily
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Deininger, P.L.; Schmid, C.W. An electron microscope study of the DNA sequence organization of the human genome. J. Mol. Biol. 1976, 106, 773–790. [Google Scholar] [CrossRef] [PubMed]
- Houck, C.M.; Rinehart, F.P.; Schmid, C.W. A ubiquitous family of repeated DNA sequences in the human genome. J. Mol. Biol. 1979, 132, 289–306. [Google Scholar] [CrossRef] [PubMed]
- Jurka, J.; Smith, T. A fundamental division in the Alu family of repeated sequences. Proc. Natl. Acad. Sci. USA 1988, 85, 4775–4778. [Google Scholar] [CrossRef] [PubMed]
- Batzer, M.A.; Deininger, P.L. A human-specific subfamily of Alu sequences. Genomics 1991, 9, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Batzer, M.A.; Kilroy, G.E.; Richard, P.E.; Shaikh, T.H.; Desselle, T.D.; Hoppens, C.L.; Deininger, P.L. Structure and variability of recently inserted Alu family members. Nucleic Acids Res. 1990, 18, 6793–6798. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.R.; Batzer, M.A.; Deininger, P.L. Evolution of the master Alu gene(s). J. Mol. Evol. 1991, 33, 311–320. [Google Scholar] [CrossRef]
- Kapitonov, V.; Jurka, J. The age of Alu subfamilies. J. Mol. Evol. 1996, 42, 59–65. [Google Scholar] [CrossRef]
- Batzer, M.A.; Stoneking, M.; Alegria-Hartman, M.; Bazan, H.; Kass, D.H.; Shaikh, T.H.; Novick, G.E.; Ioannou, P.A.; Scheer, W.D.; Herrera, R.J.; et al. African origin of human-specific polymorphic Alu insertions. Proc. Natl. Acad. Sci. USA 1994, 91, 12288–12292. [Google Scholar] [CrossRef] [PubMed]
- Konkel, M.K.; Walker, J.A.; Batzer, M.A. LINEs and SINEs of Primate Evolution. Evol. Anthropol. 2010, 19, 236–249. [Google Scholar] [CrossRef] [PubMed]
- Batzer, M.A.; Deininger, P.L.; Hellmann-Blumberg, U.; Jurka, J.; Labuda, D.; Rubin, C.M.; Schmid, C.W.; Zietkiewicz, E.; Zuckerkandl, E. Standardized nomenclature for Alu repeats. J. Mol. Evol. 1996, 42, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Bao, W.; Kojima, K.K.; Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 2015, 6, 11. [Google Scholar]
- Jurka, J. Very young Alu family from Gibbon. Repbase Rep. 2008, 8, 2248. [Google Scholar]
- Walker, J.A.; Konkel, M.K.; Ullmer, B.; Monceaux, C.P.; Ryder, O.A.; Hubley, R.; Smit, A.F.; Batzer, M.A. Orangutan Alu quiescence reveals possible source element: Support for ancient backseat drivers. Mob. DNA 2012, 3, 8. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.E.; Alkan, C.; Jiang, L.; Zhao, S.; Eichler, E.E. Comparative analysis of Alu repeats in primate genomes. Genome Res. 2009, 19, 876–885. [Google Scholar] [CrossRef]
- Ray, D.A.; Batzer, M.A. Tracking Alu evolution in New World primates. BMC Evol. Biol. 2005, 5, 51. [Google Scholar] [CrossRef]
- Perelman, P.; Johnson, W.E.; Roos, C.; Seuánez, H.N.; Horvath, J.E.; Moreira, M.A.; Kessing, B.; Pontius, J.; Roelke, M.; Rumpler, Y.; et al. A molecular phylogeny of living primates. PLoS Genet. 2011, 7, e1001342. [Google Scholar] [CrossRef]
- Smit, A.F.A.; Hubley, R.; Green, P. 2013–2015, RepeatMasker Open-4.0. 2015. Available online: http://www.repeatmasker.org (accessed on 19 October 2022).
- Baker, J.N.; Walker, J.A.; Vanchiere, J.A.; Phillippe, K.R.; St Romain, C.P.; Gonzalez-Quiroga, P.; Denham, M.W.; Mierl, J.R.; Konkel, M.K.; Batzer, M.A. Evolution of Alu Subfamily Structure in the Saimiri Lineage of New World Monkeys. Genome Biol. Evol. 2017, 9, 2365–2376. [Google Scholar] [CrossRef]
- Mao, Y.; Catacchio, C.R.; Hillier, L.W.; Porubsky, D.; Li, R.; Sulovari, A.; Fernandes, J.D.; Montinaro, F.; Gordon, D.S.; Storer, J.M.; et al. A high-quality bonobo genome refines the analysis of hominid evolution. Nature 2021, 594, 77–81. [Google Scholar] [CrossRef]
- Steely, C.J.; Baker, J.N.; Walker, J.A.; Loupe, C.D., 3rd; Batzer, M.A. Analysis of lineage-specific Alu subfamilies in the genome of the olive baboon, Papio anubis. Mob. DNA 2018, 9, 10. [Google Scholar] [CrossRef] [PubMed]
- Warren, W.C.; Harris, R.A.; Haukness, M.; Fiddes, I.T.; Murali, S.C.; Fernandes, J.; Dishuck, P.C.; Storer, J.M.; Raveendran, M.; Hillier, L.W.; et al. Sequence diversity analyses of an improved rhesus macaque genome enhance its biomedical utility. Science 2020, 370, eabc6617. [Google Scholar] [CrossRef] [PubMed]
- Worley, K.C.; Warren, W.C.; Rogers, J.; Locke, D.; Muzny, D.M.; Mardis, E.R.; Weinstock, G.M.; Tardif, S.D. The common marmoset genome provides insight into primate biology and evolution. Nat. Genet. 2014, 46, 850–857. [Google Scholar] [CrossRef]
- Kent, W.J. BLAT—The BLAST-like alignment tool. Genome Res. 2002, 12, 656–664. [Google Scholar] [CrossRef]
- Jurka, J.; Kapitonov, V.V.; Pavlicek, A.; Klonowski, P.; Kohany, O.; Walichiewicz, J. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 2005, 110, 462–467. [Google Scholar] [CrossRef]
- Orkin, J.D.; Montague, M.J.; Tejada-Martinez, D.; de Manuel, M.; Del Campo, J.; Cheves Hernandez, S.; Di Fiore, A.; Fontsere, C.; Hodgson, J.A.; Janiak, M.C.; et al. The genomics of ecological flexibility, large brains, and long lives in capuchin monkeys revealed with fecalFACS. Proc. Natl. Acad. Sci. USA 2021, 118, e2010632118. [Google Scholar] [CrossRef] [PubMed]
- Storer, J. Characterization and Amplification of Retrotransposable Elements Platy-1 and Alu in the Cebidae Lineage of Platyrrhine Primates. Doctoral Dissertations, Louisiana State University, Baton Rouge, LA, USA, 2019; p. 5053. Available online: https://digitalcommons.lsu.edu/gradschool_dissertations/5053 (accessed on 19 October 2022).
- Storer, J.M.; Walker, J.A.; Jordan, V.E.; Batzer, M.A. Sensitivity of the polyDetect computational pipeline for phylogenetic analyses. Anal. Biochem. 2020, 593, 113516. [Google Scholar] [CrossRef] [PubMed]
- Storer, J.M.; Walker, J.A.; Rewerts, L.C.; Brown, M.A.; Beckstrom, T.O.; Herke, S.W.; Roos, C.; Batzer, M.A. Owl Monkey Alu Insertion Polymorphisms and Aotus Phylogenetics. Genes 2022, 13, 2069. [Google Scholar] [CrossRef]
- Storer, J.M.; Walker, J.A.; Rockwell, C.E.; Mores, G.; Beckstrom, T.O.; Orkin, J.D.; Melin, A.D.; Phillips, K.A.; Roos, C.; Batzer, M.A. Recently Integrated Alu Elements in Capuchin Monkeys: A Resource for Cebus/Sapajus Genomics. Genes 2022, 13, 572. [Google Scholar] [CrossRef]
- Jurka, J.; Zuckerkandl, E. Free left arms as precursor molecules in the evolution of Alu sequences. J. Mol. Evol. 1991, 33, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef]
- Shingleton, A.M. Subfamily Clustering Using Label Uncertainty (for Transposable Element Families). Graduate Student Theses, Dissertations & Professional Papers. Master’s Thesis, University of Arizona, Tucson, AZ, USA, 2022; p. 11913. [Google Scholar]
- Carey, K.M.; Hubley, R.; Lesica, G.T.; Olson, D.; Roddey, J.W.; Shingleton, A.; Smit, A.F.; Wheeler, T.J. PolyA: A tool for adjudicating competing annotations of biological sequences. bioRxiv preprint. [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef]
- Lewis, P.O.; Holder, M.T.; Holsinger, K.E. Polytomies and Bayesian phylogenetic inference. Syst. Biol. 2005, 54, 241–253. [Google Scholar] [CrossRef]
- Cordaux, R.; Hedges, D.J.; Batzer, M.A. Retrotransposition of Alu elements: How many sources? Trends Genet. 2004, 20, 464–467. [Google Scholar] [CrossRef] [PubMed]
- Han, K.; Xing, J.; Wang, H.; Hedges, D.J.; Garber, R.K.; Cordaux, R.; Batzer, M.A. Under the genomic radar: The stealth model of Alu amplification. Genome Res. 2005, 15, 655–664. [Google Scholar] [CrossRef] [PubMed]
- Ray, D.A. SINEs of progress: Mobile element applications to molecular ecology. Mol. Ecol. 2007, 16, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Schneider, H.; Canavez, F.C.; Sampaio, I.; Moreira, M.A.; Tagliaro, C.H.; Seuánez, H.N. Can molecular data place each neotropical monkey in its own branch? Chromosoma 2001, 109, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Schneider, H.; Sampaio, I. The systematics and evolution of New World primates—A review. Mol. Phylogenet. Evol. 2015, 82 Pt B, 348–357. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Storer, J.M.; Walker, J.A.; Baker, J.N.; Hossain, S.; Roos, C.; Wheeler, T.J.; Batzer, M.A. Framework of the Alu Subfamily Evolution in the Platyrrhine Three-Family Clade of Cebidae, Callithrichidae, and Aotidae. Genes 2023, 14, 249. https://doi.org/10.3390/genes14020249
Storer JM, Walker JA, Baker JN, Hossain S, Roos C, Wheeler TJ, Batzer MA. Framework of the Alu Subfamily Evolution in the Platyrrhine Three-Family Clade of Cebidae, Callithrichidae, and Aotidae. Genes. 2023; 14(2):249. https://doi.org/10.3390/genes14020249
Chicago/Turabian StyleStorer, Jessica M., Jerilyn A. Walker, Jasmine N. Baker, Shifat Hossain, Christian Roos, Travis J. Wheeler, and Mark A. Batzer. 2023. "Framework of the Alu Subfamily Evolution in the Platyrrhine Three-Family Clade of Cebidae, Callithrichidae, and Aotidae" Genes 14, no. 2: 249. https://doi.org/10.3390/genes14020249