
Haploinsufficiency as a Foreground Pathomechanism of Poirer-Bienvenu Syndrome and Novel Insights Underlying the Phenotypic Continuum of CSNK2B-Associated Disorders
 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
2.2. WES, Interpretation, Validation and In Silico Functional Analysis
2.3. Plasmid Constructs
2.4. RNA Extraction from Blood
2.5. Quantitative Real-Time PCR (qRT-PCR)
2.6. Cell Cultures and Transient Transfection
2.7. Cloning PCR Product
2.8. Western Blot Analysis
2.9. Fractionation Assay
2.10. Analysis of Protein Stability
2.11. Immunocytochemical Staining
2.12. Co-Immunoprecipitation
2.13. Immunoprecipitation and Kinase Assay
2.14. Actinomycin D Treatment
3. Results
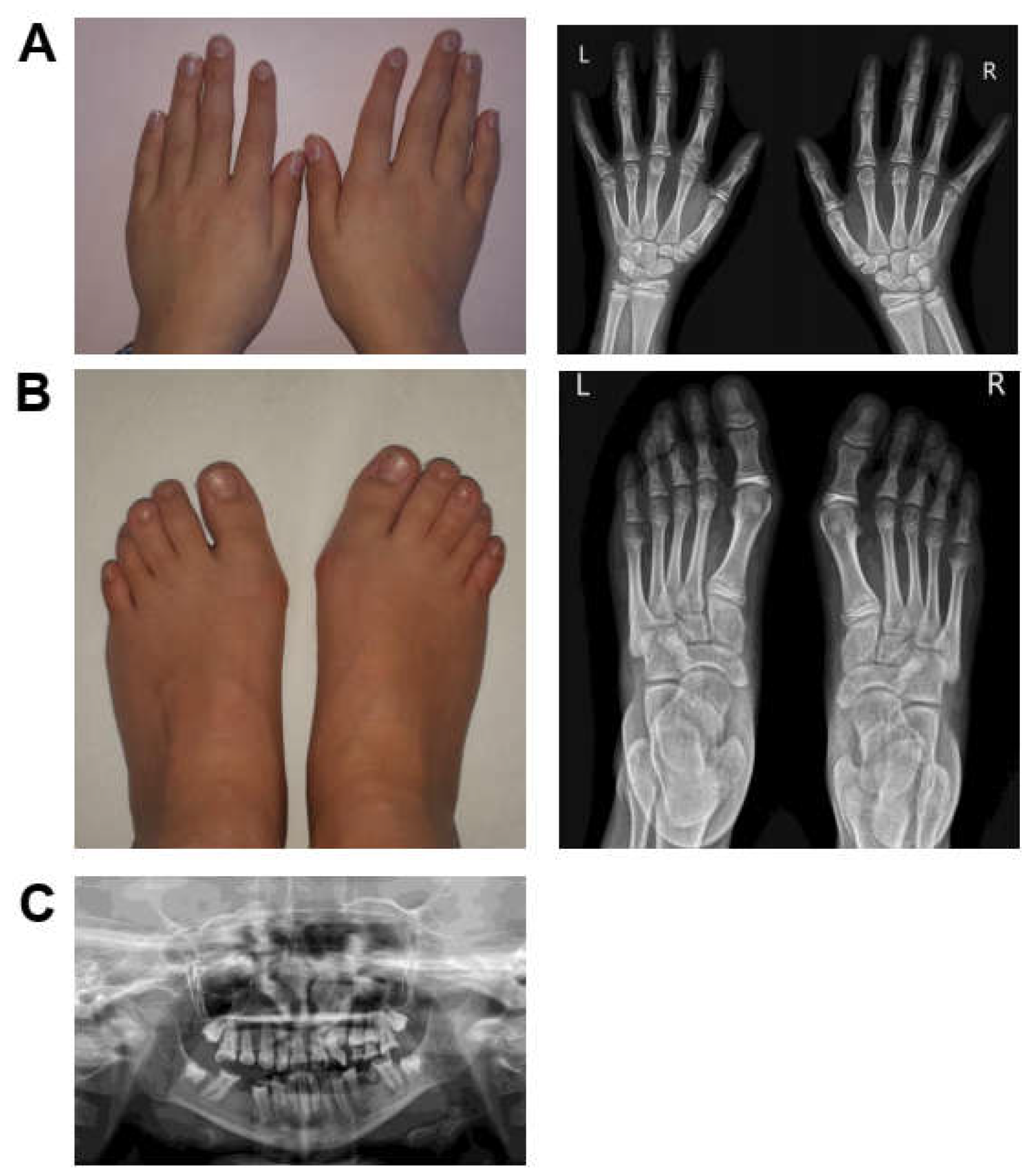
3.1. Case Presentation and Clinical Evolution
3.1.1. Patient 1
3.1.2. Patient 2
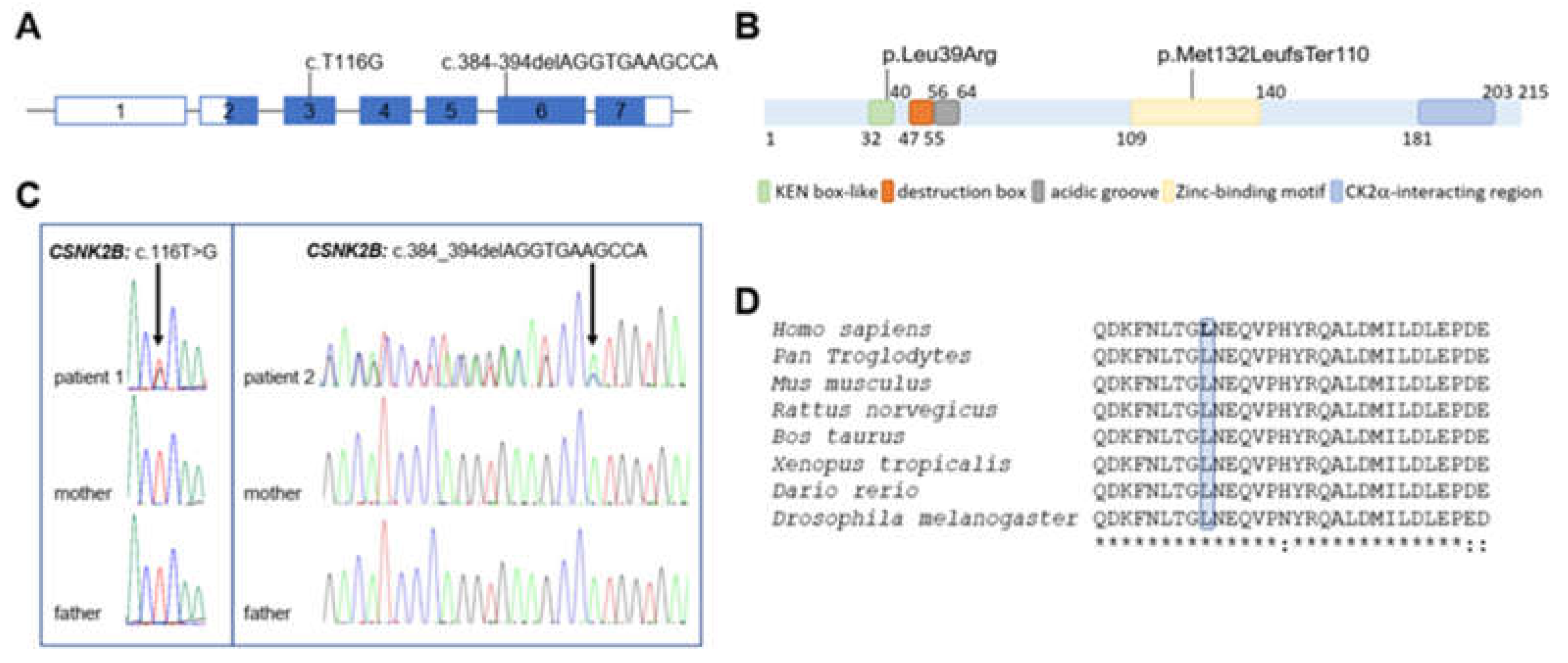
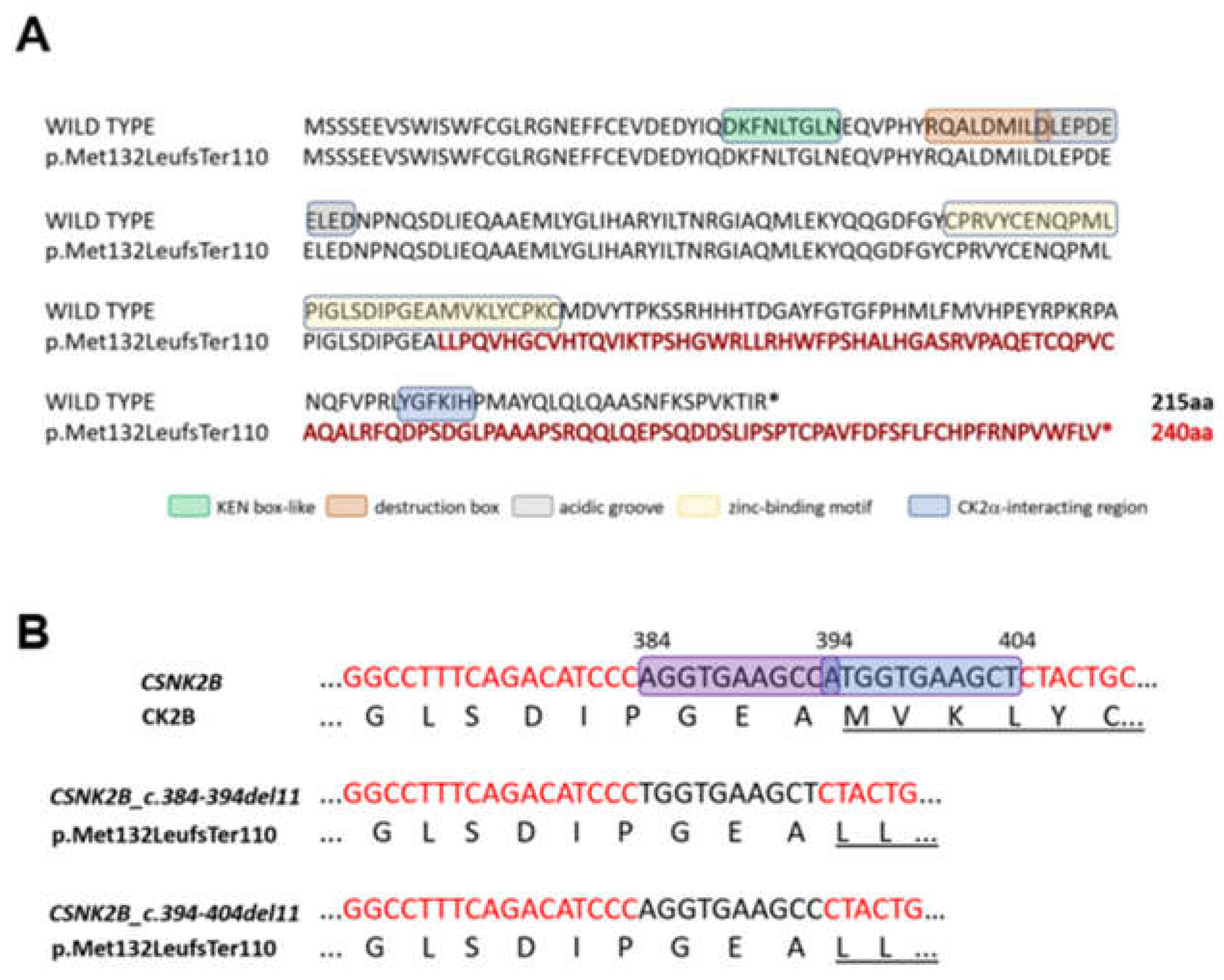
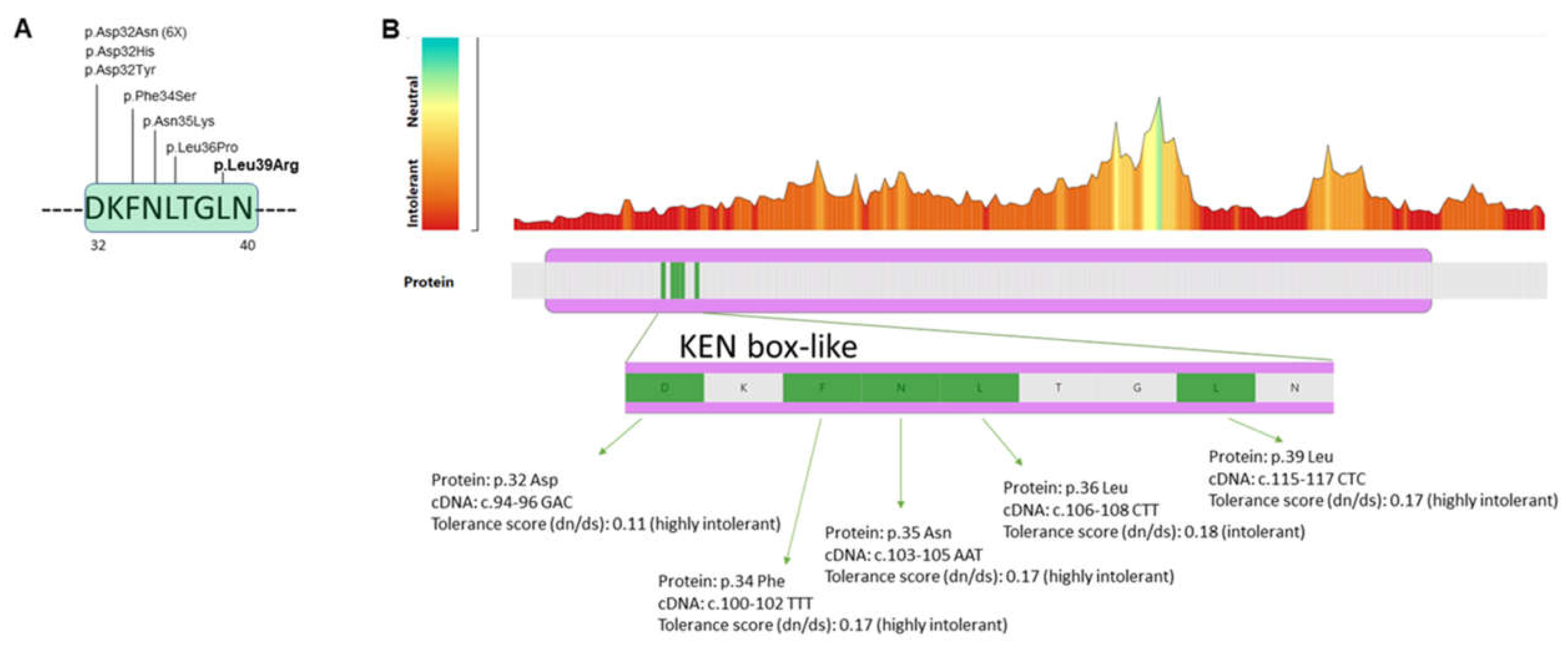
3.2. Molecular Findings
3.3. Functional Studies
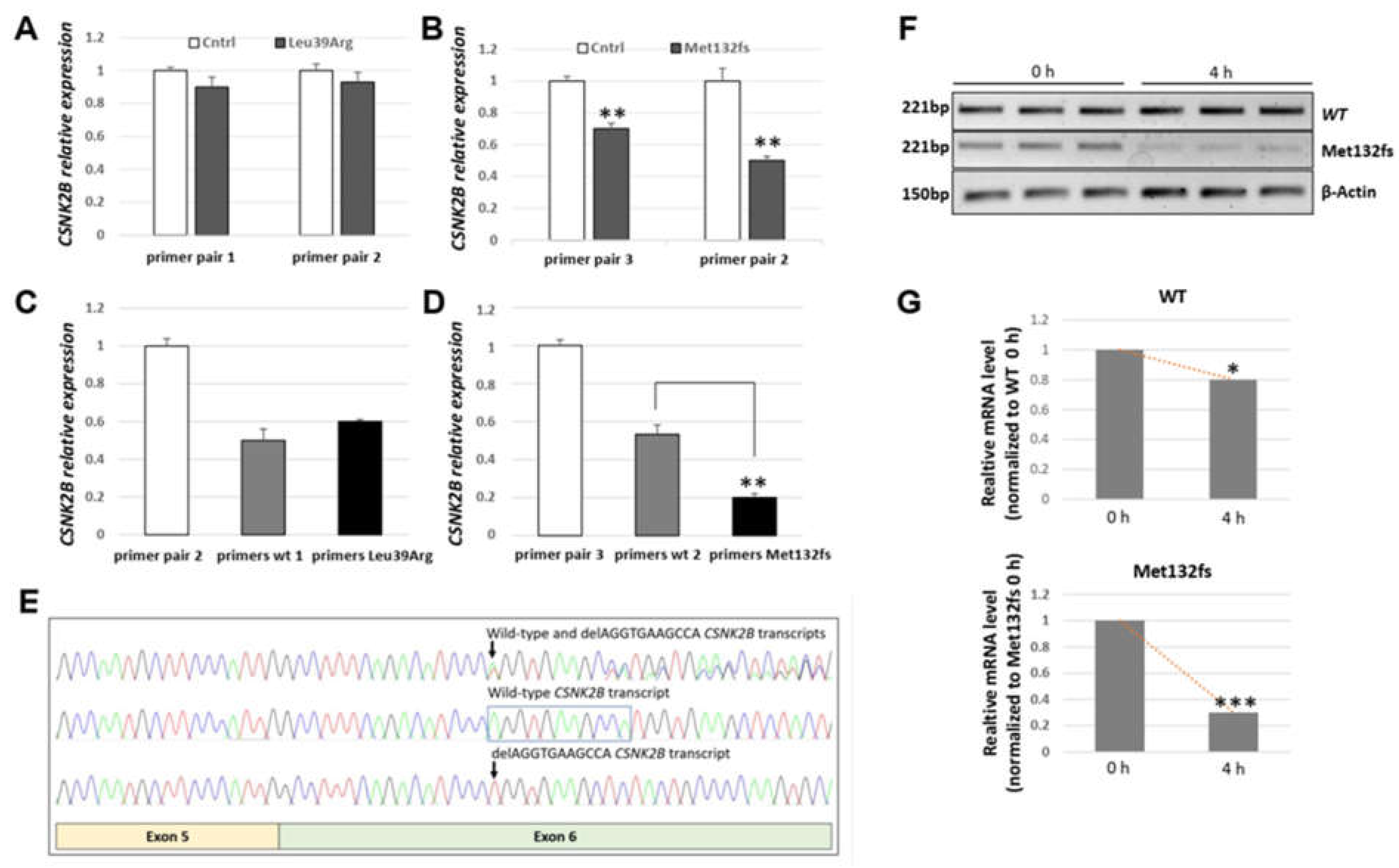
3.3.1. CSNK2B Frameshift Mutation Results in a Significantly Reduced Amount of Its mRNA
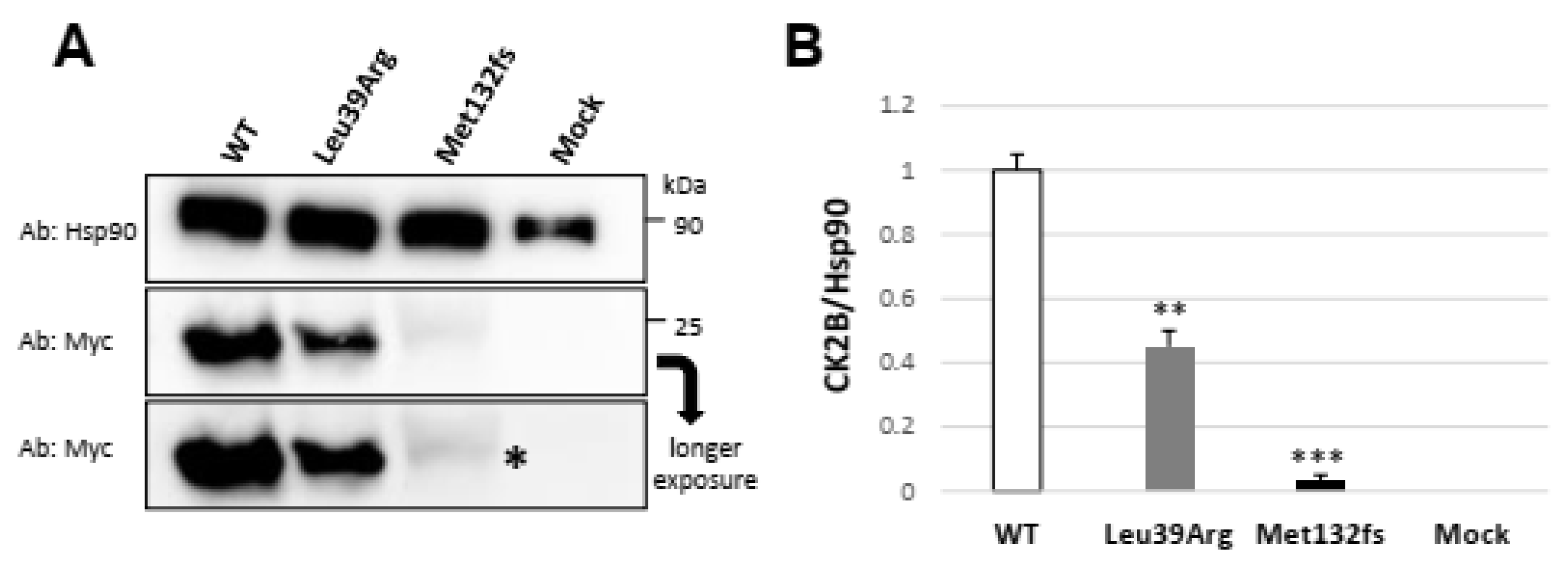
3.3.2. Transiently Expressed CSNK2B Missense Mutant Results in a Reduced CK2beta Protein Abundance
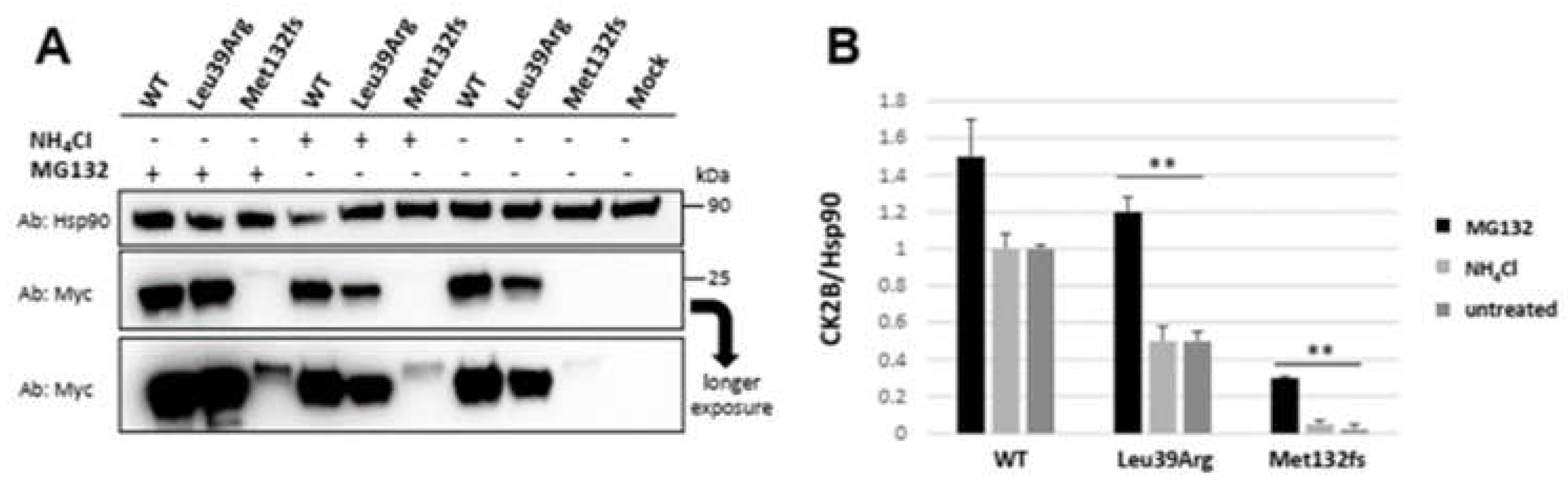
3.3.3. CSNK2B Missense Mutant Results in an Increased CK2beta Proteolysis
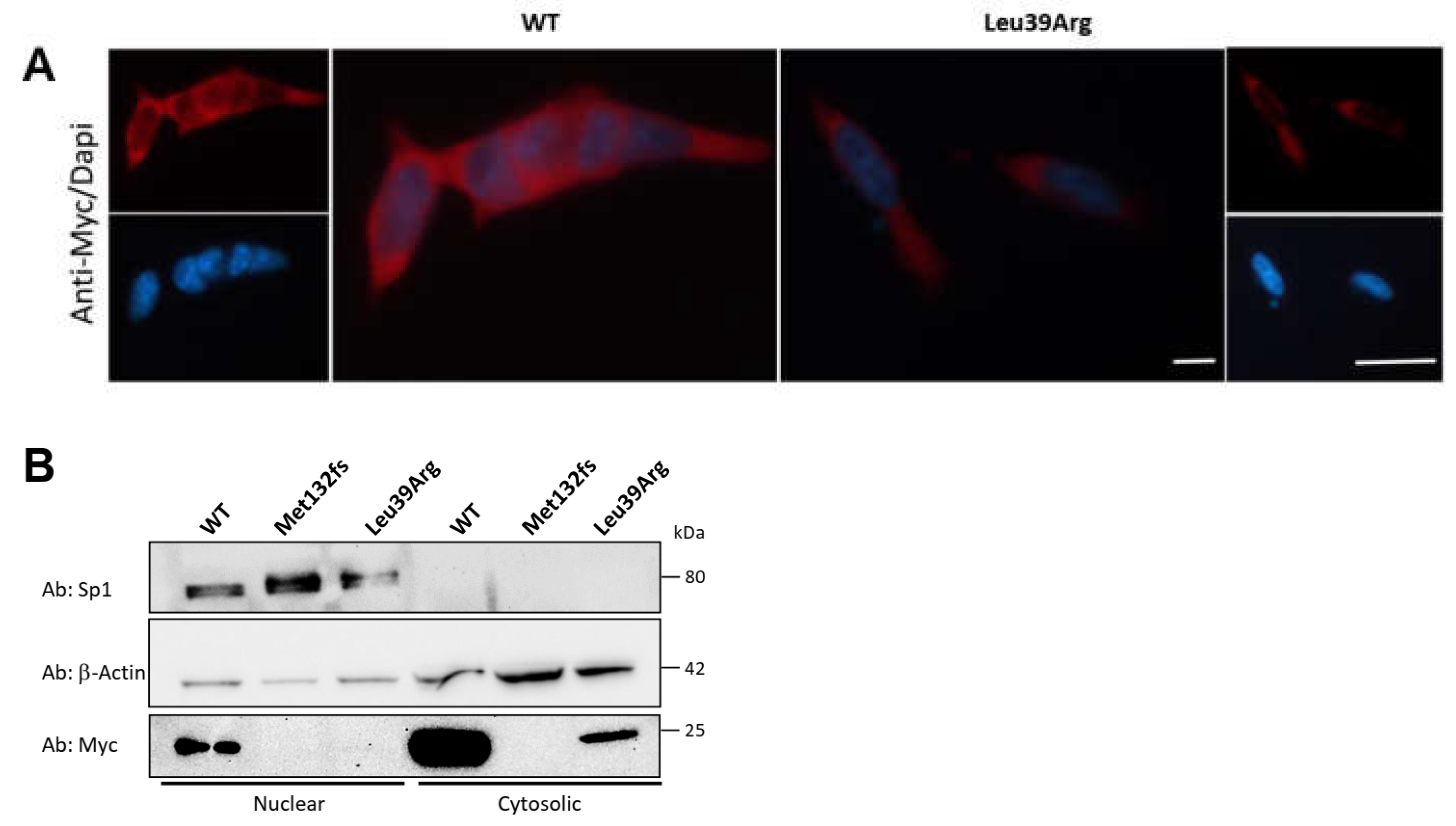
3.3.4. CK2beta Cellular Localization Is Not Impaired by Leu39Arg Mutation
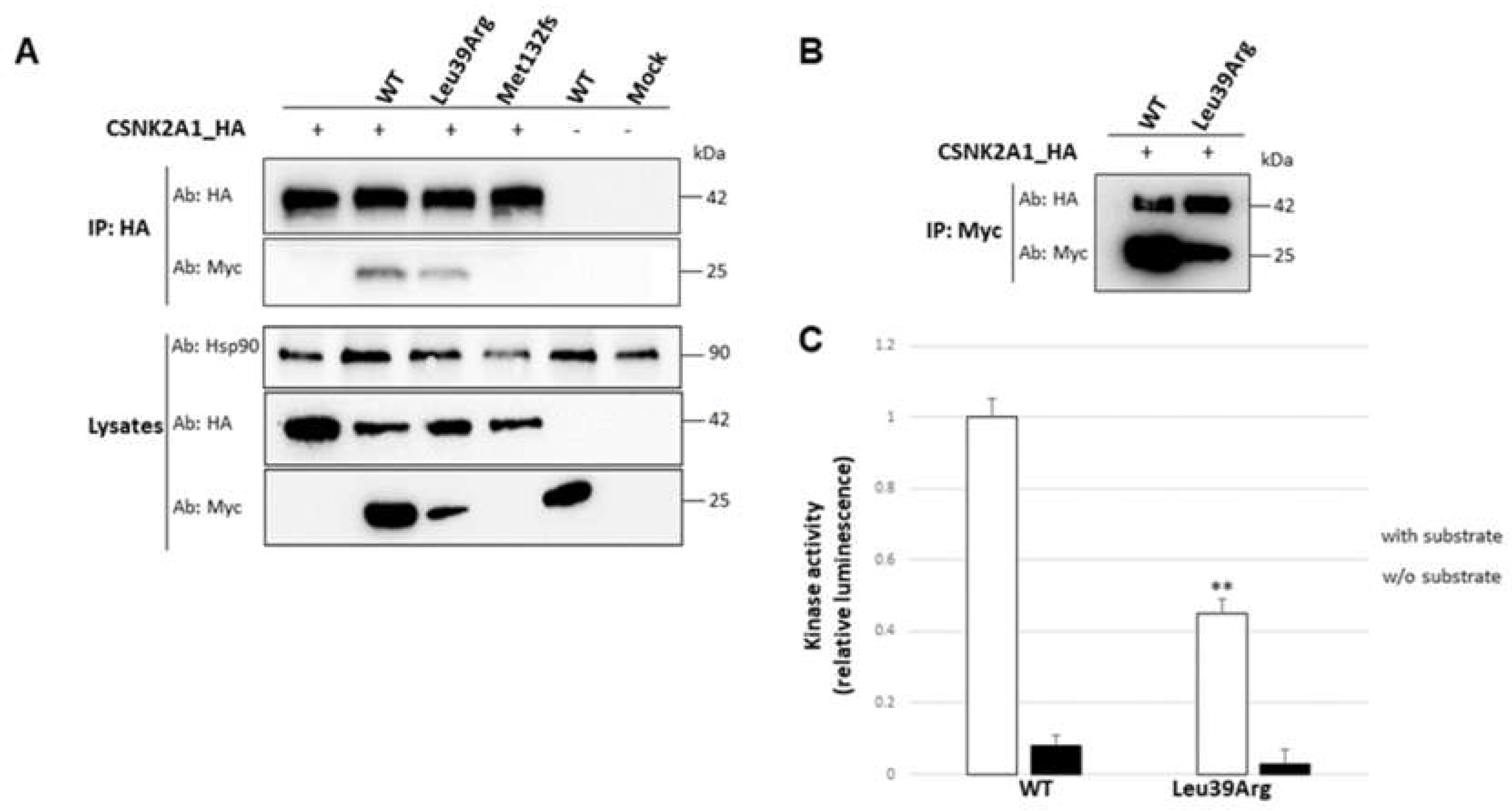
3.3.5. Interaction with the Catalytic Subunit Is Not Impaired by Leu39Arg Mutation, but the Resulting CK2 Holoenzyme Shows a Lower Kinase Activity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Niefind, K.; Guerra, B.; Ermakowa, I.; Issinger, O.G. Crystal Structure of Human Protein Kinase CK2: Insights into Basic Properties of the CK2 Holoenzyme. EMBO J. 2001, 20, 5320–5331. [Google Scholar] [CrossRef] [Green Version]
- Borgo, C.; D’Amore, C.; Sarno, S.; Salvi, M.; Ruzzene, M. Protein Kinase CK2: A Potential Therapeutic Target for Diverse Human Diseases. Signal Transduct. Target. Ther. 2021, 6, 183. [Google Scholar] [CrossRef] [PubMed]
- Pinna, L.A. Protein Kinase CK2: A Challenge to Canons. J. Cell Sci. 2002, 115 Pt 20, 3873–3878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanquet, P.R. Casein Kinase 2 as a Potentially Important Enzyme in the Nervous System. Prog. Neurobiol. 2000, 60, 211–246. [Google Scholar] [CrossRef] [PubMed]
- Chantalat, L.; Leroy, D.; Filhol, O.; Nueda, A.; Benitez, M.J.; Chambaz, E.M.; Cochet, C.; Dideberg, O. Crystal Structure of the Human Protein Kinase CK2 Regulatory Subunit Reveals Its Zinc Finger-Mediated Dimerization. EMBO J. 1999, 18, 2930–2940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilk, G.; Derksen, D.R.; Litchfield, D.W. Inducible Expression of the Regulatory Protein Kinase CK2beta Subunit: Incorporation into Complexes with Catalytic CK2 Subunits and Re-Examination of the Effects of CK2beta on Cell Proliferation. J. Cell Biochem. 2001, 84, 84–99. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Vilk, G.; Canton, D.A.; Litchfield, D.W. Phosphorylation Regulates the Stability of the Regulatory CK2beta Subunit. Oncogene 2002, 21, 3754–3764. [Google Scholar] [CrossRef] [Green Version]
- Buchou, T.; Vernet, M.; Blond, O.; Jensen, H.H.; Pointu, H.; Olsen, B.B.; Cochet, C.; Issinger, O.-G.; Boldyreff, B. Disruption of the Regulatory Beta Subunit of Protein Kinase CK2 in Mice Leads to a Cell-Autonomous Defect and Early Embryonic Lethality. Mol. Cell Biol. 2003, 23, 908–915. [Google Scholar] [CrossRef] [Green Version]
- Huillard, E.; Ziercher, L.; Blond, O.; Wong, M.; Deloulme, J.-C.; Souchelnytskyi, S.; Baudier, J.; Cochet, C.; Buchou, T. Disruption of CK2beta in Embryonic Neural Stem Cells Compromises Proliferation and Oligodendrogenesis in the Mouse Telencephalon. Mol. Cell Biol. 2010, 30, 2737–2749. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.-P.; Li, X.; Wu, Y.; Shen, Q.; Zeng, Y.; Xiong, Q.; Wei, M.; Chen, C.; Liu, J.; Huo, Y.; et al. Comprehensive Integrative Analyses Identify GLT8D1 and CSNK2B as Schizophrenia Risk Genes. Nat. Commun. 2018, 9, 838. [Google Scholar] [CrossRef]
- Poirier, K.; Hubert, L.; Viot, G.; Rio, M.; Billuart, P.; Besmond, C.; Bienvenu, T. CSNK2B Splice Site Mutations in Patients Cause Intellectual Disability with or without Myoclonic Epilepsy. Hum. Mutat. 2017, 38, 932–941. [Google Scholar] [CrossRef] [PubMed]
- Ernst, M.E.; Baugh, E.H.; Thomas, A.; Bier, L.; Lippa, N.; Stong, N.; Mulhern, M.S.; Kushary, S.; Akman, C.I.; Heinzen, E.L.; et al. CSNK2B: A Broad Spectrum of Neurodevelopmental Disability and Epilepsy Severity. Epilepsia 2021, 62, e103–e109. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Ye, F.; Chen, S.; Peng, J.; Pang, N.; Yin, F. Splicing Interruption by Intron Variants in CSNK2B Causes Poirier-Bienvenu Neurodevelopmental Syndrome: A Focus on Genotype-Phenotype Correlations. Front. Neurosci. 2022, 16, 892768. [Google Scholar] [CrossRef]
- Asif, M.; Kaygusuz, E.; Shinawi, M.; Nickelsen, A.; Hsieh, T.-C.; Wagle, P.; Budde, B.S.; Hochscherf, J.; Abdullah, U.; Höning, S.; et al. De Novo Variants of CSNK2B Cause a New Intellectual Disability-Craniodigital Syndrome by Disrupting the Canonical Wnt Signaling Pathway. HGG Adv. 2022, 3, 100111. [Google Scholar] [CrossRef]
- Köhler, S.; Gargano, M.; Matentzoglu, N.; Carmody, L.C.; Lewis-Smith, D.; Vasilevsky, N.A.; Danis, D.; Balagura, G.; Baynam, G.; Brower, A.M.; et al. The Human Phenotype Ontology in 2021. Nucleic Acids Res. 2021, 49, D1207–D1217. [Google Scholar] [CrossRef] [PubMed]
- Musante, L.; Costa, P.; Zanus, C.; Faletra, F.; Murru, F.M.; Bianco, A.M.; La Bianca, M.; Ragusa, G.; Athanasakis, E.; d’Adamo, A.P.; et al. The Genetic Diagnosis of Ultrarare DEEs: An Ongoing Challenge. Genes 2022, 13, 500. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Limongelli, I.; Marini, S.; Bellazzi, R. PaPI: Pseudo Amino Acid Composition to Score Human Protein-Coding Variants. BMC Bioinformatics 2015, 16, 123. [Google Scholar] [CrossRef] [Green Version]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting Functional Effect of Human Missense Mutations Using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 76, 7–20. [Google Scholar] [CrossRef] [Green Version]
- Sim, N.-L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT Web Server: Predicting Effects of Amino Acid Substitutions on Proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Rödelsperger, C.; Schuelke, M.; Seelow, D. MutationTaster Evaluates Disease-Causing Potential of Sequence Alterations. Nat. Methods 2010, 7, 575–576. [Google Scholar] [CrossRef] [PubMed]
- Quang, D.; Chen, Y.; Xie, X. DANN: A Deep Learning Approach for Annotating the Pathogenicity of Genetic Variants. Bioinformatics 2015, 31, 761–763. [Google Scholar] [CrossRef] [Green Version]
- Jian, X.; Boerwinkle, E.; Liu, X. In Silico Prediction of Splice-Altering Single Nucleotide Variants in the Human Genome. Nucleic Acids Res. 2014, 42, 13534–13544. [Google Scholar] [CrossRef] [Green Version]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A General Framework for Estimating the Relative Pathogenicity of Human Genetic Variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology Modelling of Protein Structures and Complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Rasband, W.S. (1997–2016) Image J. U.S. National Institute of Health, Bethesda, Maryland, USA. Available online: https://imagej.nih.gov/ij (accessed on 1 July 2022).
- Orsini, A.; Santangelo, A.; Bravin, F.; Bonuccelli, A.; Peroni, D.; Battini, R.; Foiadelli, T.; Bertini, V.; Valetto, A.; Iacomino, M.; et al. Expanding Phenotype of Poirier–Bienvenu Syndrome: New Evidence from an Italian Multicentrical Cohort of Patients. Genes 2022, 13, 276. [Google Scholar] [CrossRef]
- Bibby, A.C.; Litchfield, D.W. The Multiple Personalities of the Regulatory Subunit of Protein Kinase CK2: CK2 Dependent and CK2 Independent Roles Reveal a Secret Identity for CK2beta. Int. J. Biol. Sci. 2005, 1, 67–79. [Google Scholar] [CrossRef]
- Hsiao, M.-C.; Piotrowski, A.; Alexander, J.; Callens, T.; Fu, C.; Mikhail, F.M.; Claes, K.B.M.; Messiaen, L. Palindrome-Mediated and Replication-Dependent Pathogenic Structural Rearrangements within the NF1 Gene. Hum. Mutat. 2014, 35, 891–898. [Google Scholar] [CrossRef]
- Canton, D.A.; Zhang, C.; Litchfield, D.W. Assembly of Protein Kinase CK2: Investigation of Complex Formation between Catalytic and Regulatory Subunits Using a Zinc-Finger-Deficient Mutant of CK2beta. Biochem. J. 2001, 358 Pt 1, 87–94. [Google Scholar] [CrossRef]
- De, S.; Mühlemann, O. A Comprehensive Coverage Insurance for Cells: Revealing Links between Ribosome Collisions, Stress Responses and MRNA Surveillance. RNA Biol. 2022, 19, 609–621. [Google Scholar] [CrossRef] [PubMed]
- Powers, K.T.; Szeto, J.-Y.A.; Schaffitzel, C. New Insights into No-Go, Non-Stop and Nonsense-Mediated MRNA Decay Complexes. Curr. Opin. Struct. Biol. 2020, 65, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Zhang, Q.; Yi, S.; Qin, Z.; Shen, F.; Ou, S.; Luo, J.; He, S. De Novo CSNK2B Mutations in Five Cases of Poirier-Bienvenu Neurodevelopmental Syndrome. Front. Neurol. 2022, 13, 811092. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, M.; Tohyama, J.; Nakagawa, E.; Watanabe, Y.; Siew, C.G.; Kwong, C.S.; Yamoto, K.; Hiraide, T.; Fukuda, T.; Kaname, T.; et al. Identification of de Novo CSNK2A1 and CSNK2B Variants in Cases of Global Developmental Delay with Seizures. J. Hum. Genet. 2019, 64, 313–322. [Google Scholar] [CrossRef]
- Vissers, L.E.L.M.; van Nimwegen, K.J.M.; Schieving, J.H.; Kamsteeg, E.-J.; Kleefstra, T.; Yntema, H.G.; Pfundt, R.; van der Wilt, G.J.; Krabbenborg, L.; Brunner, H.G.; et al. A Clinical Utility Study of Exome Sequencing versus Conventional Genetic Testing in Pediatric Neurology. Genet. Med. 2017, 19, 1055–1063. [Google Scholar] [CrossRef] [Green Version]
- Turner, T.N.; Wilfert, A.B.; Bakken, T.E.; Bernier, R.A.; Pepper, M.R.; Zhang, Z.; Torene, R.I.; Retterer, K.; Eichler, E.E. Sex-Based Analysis of De Novo Variants in Neurodevelopmental Disorders. Am. J. Hum. Genet. 2019, 105, 1274–1285. [Google Scholar] [CrossRef]
- Li, J.; Gao, K.; Cai, S.; Liu, Y.; Wang, Y.; Huang, S.; Zha, J.; Hu, W.; Yu, S.; Yang, Z.; et al. Germline de Novo Variants in CSNK2B in Chinese Patients with Epilepsy. Sci. Rep. 2019, 9, 17909. [Google Scholar] [CrossRef] [Green Version]
- Hiraide, T.; Yamoto, K.; Masunaga, Y.; Asahina, M.; Endoh, Y.; Ohkubo, Y.; Matsubayashi, T.; Tsurui, S.; Yamada, H.; Yanagi, K.; et al. Genetic and Phenotypic Analysis of 101 Patients with Developmental Delay or Intellectual Disability Using Whole-Exome Sequencing. Clin. Genet. 2021, 100, 40–50. [Google Scholar] [CrossRef]
- Wilke, M.V.M.B.; Oliveira, B.M.; Pereira, A.; Doriqui, M.J.R.; Kok, F.; Souza, C.F.M. Two Different Presentations of de Novo Variants of CSNK2B: Two Case Reports. J. Med. Case Rep. 2022, 16, 4. [Google Scholar] [CrossRef]
- Unni, P.; Friend, J.; Weinberg, J.; Okur, V.; Hochscherf, J.; Dominguez, I. Predictive Functional, Statistical and Structural Analysis of CSNK2A1 and CSNK2B Variants Linked to Neurodevelopmental Diseases. Front. Mol. Biosci. 2022, 9, 851547. [Google Scholar] [CrossRef]
- Wiel, L.; Baakman, C.; Gilissen, D.; Veltman, J.A.; Vriend, G.; Gilissen, C. MetaDome: Pathogenicity Analysis of Genetic Variants through Aggregation of Homologous Human Protein Domains. Hum. Mutat. 2019, 40, 1030–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glotzer, M.; Murray, A.W.; Kirschner, M.W. Cyclin Is Degraded by the Ubiquitin Pathway. Nature 1991, 349, 132–138. [Google Scholar] [CrossRef] [PubMed]
- King, R.W.; Glotzer, M.; Kirschner, M.W. Mutagenic Analysis of the Destruction Signal of Mitotic Cyclins and Structural Characterization of Ubiquitinated Intermediates. Mol. Biol. Cell 1996, 7, 1343–1357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borgo, C.; D’Amore, C.; Cesaro, L.; Sarno, S.; Pinna, L.A.; Ruzzene, M.; Salvi, M. How Can a Traffic Light Properly Work If It Is Always Green? The Paradox of CK2 Signaling. Crit. Rev. Biochem. Mol. Biol. 2021, 56, 321–359. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CK2B Mutation (NP_001311.3) | p.Asp32Asn | p.Asp32His | p.Asp32Tyr | p.Phe34ser | p.Asn35Lys | p.Leu36Pro | p.Leu39Arg |
|---|---|---|---|---|---|---|---|
| CADD score | 28.9 | 29.7 | 32 | 29.4 | 23.8 | 28.9 | 29.5 |
| I-Mutant2.0 | ΔΔG= −0.49 | ΔΔG= −0.97 | ΔΔG= −0.28 | ΔΔG= −2.74 | ΔΔG= −2.42 | ΔΔG= −2.55 | ΔΔG= −2.32 |
| SDM | ΔΔG= −0.21 | ΔΔG= −0.01 | ΔΔG= −0.05 | ΔΔG= −0.39 | ΔΔG= −0.98 | ΔΔG= −2.29 | ΔΔG= −2.67 |
| Reference | Patient 2 [14] | Patient [14] | Patient 7 [14] Previously Patient 25 [12] | Patient 8 [12] | Patient 14 [12] | Patient 2691 [38] | Patient 1 [14] | Patient 2 [39] | Patient 10 [12] | Patient 12 [12] | Patient 48 [35] | Patient 1 Current Study | Total |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mutation (NP_001311.3) | p.Asp32Asn | p.Asp32Asn | p.Asp32Asn | p.Asp32Asn | p.Asp32Asn | p.Asp32Asn | p.Asp32His | p.Asp32Tyr | p.Phe34Ser | p.Asn35Lys | p.Leu36Pro | p.Leu39Arg | |
| Gender | F | F | M | M | M | M | F | M | F | M | M | F | 5F:7M |
| Age (years; months) | 10y | 2y | 13y | 12y | 31y | 2y (died) | 19y | 5y | 17y | 19m | 11y 5m | 11y | 19m-19y |
| Head abnormalities | - | - | - | NA | NA | microcephaly (HP:0000252) | - | Microbrachy-cephaly | - | macrocephaly (HP:0000256) | NA | microcephaly (HP:0000252) | 4/9 (44.4%) |
| Craniofacial features | asymmetrical ears (HP:0010722); deep-set eyes (HP:0000490); hypoplastic alae nasi (HP:0000430); thin upper lip (HP:0000219); prognathism (HP:0000303); pointed chin (HP:0000307) | deep-set eyes (HP:0000490); hypoplastic alae nasi (HP:0000430); prognathism (HP:0000303) | anteverted ears (HP:0001249); upslantong palpebral fissures (HP:0000582); bulbous nose (HP:0000414); smooth philtrum (HP:0000319); thin upper lip vermilion (HP:0000219) | mild dysmorphic features | mild dysmorphic features | External ear anomaly (HP:0000356); thick eyebrows (HP:0000574); short neck (HP:0000470) | small ears (HP:0008551) with prominent antitragus; deep-set eyes (HP:0000490); broad nasal bridge (HP:0000431); hypoplastic alae nasi (HP:0000430); thin upper lip (HP:0000219); prognathism (HP:0000303); pointed chin (HP:0000307) | prominent and dyplastic ears (HP:0000411; HP:0000377); low nasal bridge (HP:0005280); anteverted nares (HP:0000463); malar hypoplasia (HP:0000272); high and narrow palate (HP:0002705) | mild dysmorphic features | mild dysmorphic features | NA | overfolded helix (HP:0000396); broad forehead (HP:0000337); frontal bossing (HP:0002007); sparse eyebrows (HP:0045075), hypertelorism (HP:0000316); depressed nasal bridge (HP:0005280); prognathism (HP:0000303); pointed chin (HP:0000307) | 11/11 (100%) |
| Ectodermal anomalies | thin hair (HP:0008070); small teeth (HP:0000691) | sparse hair (HP:0008070); coarse hair (HP:0002208); delayed teeth eruption (HP:0000684) | supernumerary tooth (HP:0011069) | NA | NA | - | thin hair (HP:0008070); hypodontia (HP:000668) | duplicate upper right incisor (HP:0011064); nail hypoplasia (HP:0001792) | NA | NA | NA | thin hair (HP:0008070); delayed teeth eruption (HP:0000684); teeth agenesis (HP:0009804) | 6/7 (85.7%) |
| Skeletal abnormalities | tapered fingers (HP:0001182); bilateral clinodactyly of toes (HP:0001863) | brachydactyly (HP:0001156); contractures of the hand (HP:0009473) | brachydactyly (HP:0001156) | NA | NA | - | bilateral syndactily of the fingers (HP:0010492) and toes (HP:0001770); clinodactyly (HP:0001863); broad thumb (HP:0011304) | short fifth fingers (HP:0009237); tapered distal phalanges of fingers (HP:0009884); left hand postaxial polydactyly (HP:0100259) | unilateral polydactily (HP:0010442) | NA | NA | tapered fingers (HP:0001182); hallux valgus (HP:0001822); butterfly vertebrae (HP:0003316) | 7/8 (87.5%) |
| Short stature (HP:0004322) | - | - | - | NA | NA | + | + | NA | NA | NA | NA | + | 3/6 (50%) |
| Developmental Delay (HP:0001263) | + | + | + | + | + | + | + | + | + | + | NA | + | 11/11 (100%) |
| Intellectual disability (HP:0001249) | + | NA | mild-moderate | moderate (HP:0002342) | moderate (HP:0002342) | mild (HP:0001256) | moderate (HP:0002342) | NA | mild (HP:0001256) | NA | NA | mild (HP:0001256) | 8/8 (100%) |
| Speech impairment (HP:0002167) | + | + | dyspraxia (HP:0011098) | + ( 25 words at 5) | + (few words) | + | dysarthria (HP:0001260) | NA | delayed speech (HP:0000750) | does not babble | NA | Delayed speech and language development (HP:0000750); abnormal verbal communicative behavior (HP: 4000072) | 10/10 (100%) |
| Seizures (HP:0001250) | - (EEG abnormalities) | - | + (absences; onset: 7y) | + (febrile sz; onset: 1.5m) | + (absences, tonic-spasms; onset: 2y) | - | + (tonic-clonic; onset: NA) | + (onset: 9 m) muscular tone loss abnormal eyes movements | - | - | + (onset: NA) | + (absences; onset: 5y) | 7/12 (58.3%) |
| Abnormal brain MRI | NA | NA | - | (2y) mild to moderate diffuse abnormal signal in the cerebral white matter (HP:0002500); low volume ventral pons | NA | - | NA | NA | periventricular gliosis | NA | NA | Chiari malformation (HP:0002308); syringomyelia (HP:0003396) | 3/5 (60%) |
| Cardiac/ vascular abnormalities | WPW (HP:0001716) | fenestrated atrial septal defect (HP:0001631) | patent ductus arteriosus (HP:0001643) | NA | NA | - | Ebstein’s anomaly (HP:0010316) atrial septal defect (HP:0001631) | NA | NA | patent foramen ovale (HP:0001655) | NA | arteriovenous vascular abnormality (HP:0100026) | 6/7 (85.7%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Stazio, M.; Zanus, C.; Faletra, F.; Pesaresi, A.; Ziccardi, I.; Morgan, A.; Girotto, G.; Costa, P.; Carrozzi, M.; d’Adamo, A.P.; et al. Haploinsufficiency as a Foreground Pathomechanism of Poirer-Bienvenu Syndrome and Novel Insights Underlying the Phenotypic Continuum of CSNK2B-Associated Disorders. Genes 2023, 14, 250. https://doi.org/10.3390/genes14020250
Di Stazio M, Zanus C, Faletra F, Pesaresi A, Ziccardi I, Morgan A, Girotto G, Costa P, Carrozzi M, d’Adamo AP, et al. Haploinsufficiency as a Foreground Pathomechanism of Poirer-Bienvenu Syndrome and Novel Insights Underlying the Phenotypic Continuum of CSNK2B-Associated Disorders. Genes. 2023; 14(2):250. https://doi.org/10.3390/genes14020250
Chicago/Turabian StyleDi Stazio, Mariateresa, Caterina Zanus, Flavio Faletra, Alessia Pesaresi, Ilaria Ziccardi, Anna Morgan, Giorgia Girotto, Paola Costa, Marco Carrozzi, Adamo P. d’Adamo, and et al. 2023. "Haploinsufficiency as a Foreground Pathomechanism of Poirer-Bienvenu Syndrome and Novel Insights Underlying the Phenotypic Continuum of CSNK2B-Associated Disorders" Genes 14, no. 2: 250. https://doi.org/10.3390/genes14020250