
Gene Expression and Epigenetic Regulation in the Prefrontal Cortex of Schizophrenia

{kind=link}
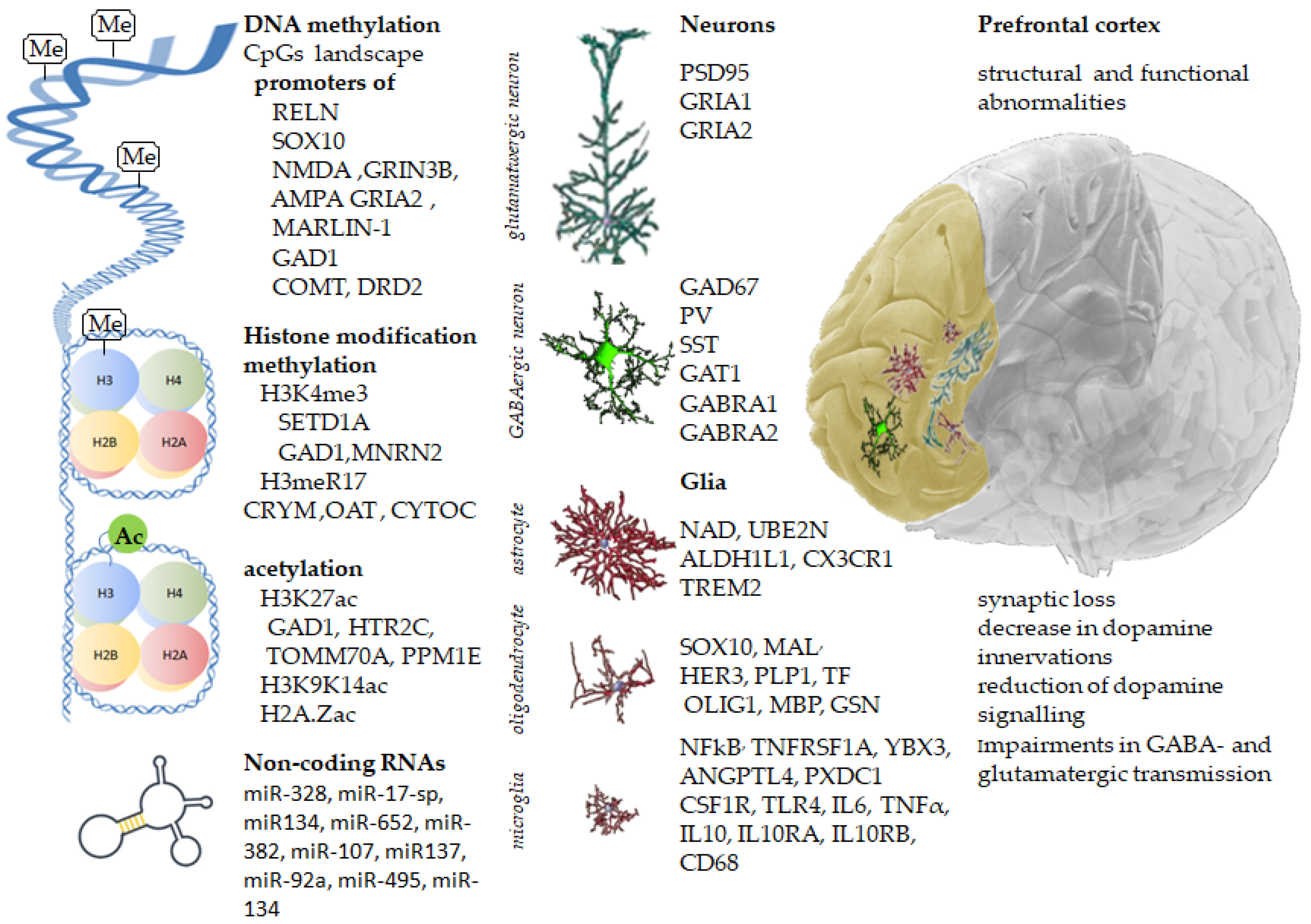
Abstract
:1. Prefrontal Cortex Development and Function
2. Prefrontal Cortical Pathology in Schizophrenia
3. Genetic Background of Schizophrenia
4. Abnormalities in Cortical Gene Expression in Schizophrenia
4.1. Glutamate-Related Genes
4.2. GABA-Related Genes
4.3. Dopamine-Related Genes
4.4. Plasticity-Related Genes
4.5. Myelination-Related Genes
4.6. Metabolic-Related Genes
4.7. Inflammation-Related Genes
5. Epigenetic Regulation
5.1. DNA Methylation
5.2. Histone Modifications
5.2.1. Histone Methylation
5.2.2. Histone Acetylation
5.3. Non-Coding RNAs
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Carlen, M. What constitutes the prefrontal cortex? Science 2017, 358, 478–482. [Google Scholar] [CrossRef] [Green Version]
- Sakurai, T.; Gamo, N.J.; Hikida, T.; Kim, S.H.; Murai, T.; Tomoda, T.; Sawa, A. Converging models of schizophrenia--Network alterations of prefrontal cortex underlying cognitive impairments. Prog. Neurobiol 2015, 134, 178–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsubomoto, M.; Kawabata, R.; Zhu, X.; Minabe, Y.; Chen, K.; Lewis, D.A.; Hashimoto, T. Expression of Transcripts Selective for GABA Neuron Subpopulations across the Cortical Visuospatial Working Memory Network in the Healthy State and Schizophrenia. Cereb. Cortex 2019, 29, 3540–3550. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.S.; Mack, N.R.; Shu, Y.; Gao, W.J. Prefrontal GABAergic Interneurons Gate Long-Range Afferents to Regulate Prefrontal Cortex-Associated Complex Behaviors. Front. Neural Circuits 2021, 15, 716408. [Google Scholar] [CrossRef] [PubMed]
- Dienel, S.J.; Lewis, D.A. Alterations in cortical interneurons and cognitive function in schizophrenia. Neurobiol. Dis. 2019, 131, 104208. [Google Scholar] [CrossRef]
- Ferguson, B.R.; Gao, W.-J. PV Interneurons: Critical Regulators of E/I Balance for Prefrontal Cortex-Dependent Behavior and Psychiatric Disorders. Front. Neural Circuits 2018, 12, 37. [Google Scholar] [CrossRef] [Green Version]
- Tremblay, R.; Lee, S.; Rudy, B. GABAergic interneurons in the neocortex: From cellular properties to circuits. Neuron 2016, 91, 260–292. [Google Scholar] [CrossRef] [Green Version]
- Selemon, L.D.; Zecevic, N. Schizophrenia: A tale of two critical periods for prefrontal cortical development. Transl. Psychiatry 2015, 5, e623. [Google Scholar] [CrossRef] [Green Version]
- Sakurai, T.; Gamo, N.J. Cognitive functions associated with developing prefrontal cortex during adolescence and developmental neuropsychiatric disorders. Neurobiol. Dis. 2018, 131, 104322. [Google Scholar] [CrossRef]
- Bennett, M.R. Synapse formation and regression in the cortex during adolescence and in schizophrenia. Med. J. Aust. 2009, 190, S14–S16. [Google Scholar] [CrossRef]
- Chini, M.; Hanganu-Opatz, I.L. Prefrontal Cortex Development in Health and Disease: Lessons from Rodents and Humans. Trends Neurosci. 2020, 44, 227–240. [Google Scholar] [CrossRef] [PubMed]
- Sommer, I.E.; Bearden, C.E.; van Dellen, E.; Breetvelt, E.J.; Duijff, S.N.; Maijer, K.; van Amelsvoort, T.; de Haan, L.; Gur, R.E.; Arango, C.; et al. Early interventions in risk groups for schizophrenia: What are we waiting for? NPJ Schizophr. 2016, 2, 16003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, V.; Nichenmetla, S.; Chhabra, H.; Sreeraj, V.S.; Rao, N.P.; Kesavan, M.; Varambally, S.; Venkatasubramanian, G.; Gangadhar, B.N. Prefrontal cortex activation during working memory task in schizophrenia: A fNIRS study. Asian J. Psychiatry 2020, 56, 102507. [Google Scholar] [CrossRef]
- Yan, Z.; Rein, B. Mechanisms of synaptic transmission dysregulation in the prefrontal cortex: Pathophysiological implications. Mol. Psychiatry 2021, 27, 445–465. [Google Scholar] [CrossRef] [PubMed]
- Mirnics, K.; Middleton, F.A.; Marquez, A.; Lewis, D.A.; Levitt, P. Molecular Characterization of Schizophrenia Viewed by Microarray Analysis of Gene Expression in Prefrontal Cortex. Neuron 2000, 28, 53–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selemon, L.D.; Goldman-Rakic, P.S. The reduced neuropil hypothesis: A circuit based model of schizophrenia. Biol. Psychiatry 1999, 45, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Thune, J.J.; Uylings, H.B.; Pakkenberg, B. No deficit in total number of neurons in the prefrontal cortex in schizophrenics. J. Psychiatr. Res. 2001, 35, 15–21. [Google Scholar] [CrossRef]
- Mauney, S.A.; Athanas, K.M.; Pantazopoulos, H.; Shaskan, N.; Passeri, E.; Berretta, S.; Woo, T.-U.W. Developmental Pattern of Perineuronal Nets in the Human Prefrontal Cortex and Their Deficit in Schizophrenia. Biol. Psychiatry 2013, 74, 427–435. [Google Scholar] [CrossRef] [Green Version]
- Kristiansen, L.; Huerta, I.; Beneyto, M.; Meador-Woodruff, J.H. NMDA receptors and schizophrenia. Curr. Opin. Pharmacol. 2007, 7, 48–55. [Google Scholar] [CrossRef]
- Smucny, J.; Carter, C.S.; Maddock, R.J. Medial Prefrontal Cortex Glutamate Is Reduced in Schizophrenia and Moderated by Measurement Quality: A Meta-analysis of Proton Magnetic Resonance Spectroscopy Studies. Biol. Psychiatry 2021, 90, 643–651. [Google Scholar] [CrossRef]
- Akil, M.; Pierri, J.N.; Whitehead, R.E.; Edgar, C.L.; Mohila, C.; Sampson, A.R.; Lewis, D.A. Lamina-Specific Alterations in the Dopamine Innervation of the Prefrontal Cortex in Schizophrenic Subjects. Am. J. Psychiatry 1999, 156, 1580–1589. [Google Scholar] [CrossRef] [PubMed]
- Abi-Dargham, A.; Mawlawi, O.; Lombardo, I.; Gil, R.; Martinez, D.; Huang, Y.; Hwang, D.-R.; Keilp, J.; Kochan, L.; Van Heertum, R.; et al. Prefrontal Dopamine D1Receptors and Working Memory in Schizophrenia. J. Neurosci. 2002, 22, 3708–3719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, P.J.; Weinberger, D.R. Schizophrenia genes, gene expression, and neuropathology: On the matter of their convergence. Mol. Psychiatry 2004, 10, 40–68. [Google Scholar] [CrossRef] [Green Version]
- Cariaga-Martinez, A.; Saiz-Ruiz, J.; Alelú-Paz, R. From Linkage Studies to Epigenetics: What We Know and What We Need to Know in the Neurobiology of Schizophrenia. Front. Neurosci. 2016, 10, 202. [Google Scholar] [CrossRef] [Green Version]
- Madzarac, Z.; Tudor, L.; Sagud, M.; Erjavec, G.N.; Peles, A.M.; Pivac, N. The Associations between COMT and MAO-B Genetic Variants with Negative Symptoms in Patients with Schizophrenia. Curr. Issues Mol. Biol. 2021, 43, 618–636. [Google Scholar] [CrossRef]
- Williams, N.M.; Preece, A.; Morris, D.W.; Spurlock, G.; Bray, N.J.; Stephens, M.; Norton, N.; Williams, H.; Clement, M.; Dwyer, S.; et al. Identification in 2 Independent Samples of a Novel Schizophrenia RiskHaplotype of the Dystrobrevin Binding Protein Gene (DTNBP1). Arch. Gen. Psychiatry 2004, 61, 336–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donohoe, G.; Frodl, T.; Morris, D.; Spoletini, I.; Cannon, D.; Cherubini, A.; Caltagirone, C.; Bossù, P.; McDonald, C.; Gill, M.; et al. Reduced Occipital and Prefrontal Brain Volumes in Dysbindin-Associated Schizophrenia. Neuropsychopharmacology 2009, 35, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, M.; Takahashi, A.; Kamatani, Y.; Momozawa, Y.; Saito, T.; Kondo, K.; Shimasaki, A.; Kawase, K.; Sakusabe, T.; Iwayama, Y.; et al. Genome-Wide Association Study Detected Novel Susceptibility Genes for Schizophrenia and Shared Trans-Populations/Diseases Genetic Effect. Schizophr. Bull. 2018, 45, 824–834. [Google Scholar] [CrossRef] [PubMed]
- Paul, A.; Nawalpuri, B.; Shah, D.; Sateesh, S.; Muddashetty, R.S.; Clement, J. Differential Regulation of Syngap1 Translation by FMRP Modulates eEF2 Mediated Response on NMDAR Activity. Front. Mol. Neurosci. 2019, 12, 97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; He, M.; Tian, X.; Guo, Y.; Liu, F.; Li, Y.; Zhang, H.; Lu, X.; Xu, D.; Zhou, R.; et al. Transgenic overexpression of furin increases epileptic susceptibility. Cell Death Dis. 2018, 9, 1058. [Google Scholar] [CrossRef]
- Yu, H.; Yan, H.; Li, J.; Li, Z.; Zhang, X.; Ma, Y.; Mei, L.; Liu, C.; Cai, L.; Wang, Q.; et al. Common variants on 2p16.1, 6p22.1 and 10q24.32 are associated with schizophrenia in Han Chinese population. Mol. Psychiatry 2016, 22, 954–960. [Google Scholar] [CrossRef] [PubMed]
- Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014, 511, 421–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fromer, M.; Pocklington, A.J.; Kavanagh, D.H.; Williams, H.J.; Dwyer, S.; Gormley, P.; Georgieva, L.; Rees, E.; Palta, P.; Ruderfer, D.M.; et al. De novo mutations in schizophrenia implicate synaptic networks. Nature 2014, 506, 179–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirov, G.; Pocklington, A.; Holmans, P.; Ivanov, D.; Ikeda, M.; Ruderfer, D.; Moran, J.; Chambert, K.; Toncheva, D.; Georgieva, L.; et al. De novo CNV analysis implicates specific abnormalities of postsynaptic signalling complexes in the pathogenesis of schizophrenia. Mol. Psychiatry 2011, 17, 142–153. [Google Scholar] [CrossRef] [Green Version]
- Purcell, S.M.; Moran, J.L.; Fromer, M.; Ruderfer, D.; Solovieff, N.; Roussos, P.; O’Dushlaine, C.; Chambert, K.; Bergen, S.E.; Kähler, A.; et al. A polygenic burden of rare disruptive mutations in schizophrenia. Nature 2014, 506, 185–190. [Google Scholar] [CrossRef] [Green Version]
- The Network and Pathway Analysis Subgroup of the Psychiatric Genomics Consortium. Psychiatric genome-wide association study analyses implicate neuronal, immune and histone pathways. Nat Neurosci 2015, 18, 199–209. [Google Scholar] [CrossRef]
- Liu, H.; Xu, L.; Fu, J.; Su, Q.; Liu, N.; Xu, J.; Tang, J.; Li, W.; Zhao, F.; Ding, H.; et al. Prefrontal Granule Cell-Related Genes and Schizophrenia. Cereb. Cortex 2020, 31, 2268–2277. [Google Scholar] [CrossRef]
- Stahl, S.M. Beyond the dopamine hypothesis of schizophrenia to three neural networks of psychosis: Dopamine, serotonin, and glutamate. CNS Spectrums 2018, 23, 187–191. [Google Scholar] [CrossRef] [Green Version]
- Weickert, C.S.; Fung, S.J.; Catts, V.S.; Schofield, P.R.; Allen, K.M.; Moore, L.T.; Newell, K.A.; Pellen, D.; Huang, X.-F.; Catts, S.V.; et al. Molecular evidence of N-methyl-D-aspartate receptor hypofunction in schizophrenia. Mol. Psychiatry 2013, 18, 1185–1192. [Google Scholar] [CrossRef] [Green Version]
- Rahman, T.; Purves-Tyson, T.; Geddes, A.E.; Huang, X.F.; Newell, K.A.; Weickert, C.S. N-Methyl-d-Aspartate receptor and inflammation in dorsolateral prefrontal cortex in schizophrenia. Schizophr. Res. 2022, 240, 61–70. [Google Scholar] [CrossRef]
- Beneyto, M.; Meador-Woodruff, J.H. Lamina-Specific Abnormalities of NMDA Receptor-Associated Postsynaptic Protein Transcripts in the Prefrontal Cortex in Schizophrenia and Bipolar Disorder. Neuropsychopharmacology 2007, 33, 2175–2186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaffe, A.E.; The BrainSeq Consortium; Straub, R.E.; Shin, J.H.; Tao, R.; Gao, Y.; Collado-Torres, L.; Kam-Thong, T.; Xi, H.S.; Quan, J.; et al. Developmental and genetic regulation of the human cortex transcriptome illuminate schizophrenia pathogenesis. Nat. Neurosci. 2018, 21, 1117–1125. [Google Scholar] [CrossRef] [PubMed]
- Kristiansen, L.V.; Beneyto, M.; Haroutunian, V.; Meador-Woodruff, J.H. Changes in NMDA receptor subunits and interacting PSD proteins in dorsolateral prefrontal and anterior cingulate cortex indicate abnormal regional expression in schizophrenia. Mol. Psychiatry 2006, 11, 737–747. [Google Scholar] [CrossRef]
- Ohnuma, T.; Kato, H.; Arai, H.; Faull, R.L.M.; McKenna, P.J.; Emson, P.C. Gene expression of PSD95 in prefrontal cortex and hippocampus in schizophrenia. Neuroreport 2000, 11, 3133–3137. [Google Scholar] [CrossRef]
- Vawter, M.P.; Crook, J.M.; Hyde, T.M.; Kleinman, J.E.; Weinberger, D.R.; Becker, K.G.; Freed, W.J. Microarray analysis of gene expression in the prefrontal cortex in schizophrenia: A preliminary study. Schizophr. Res. 2002, 58, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Yonezawa, K.; Tani, H.; Nakajima, S.; Nagai, N.; Koizumi, T.; Miyazaki, T.; Mimura, M.; Takahashi, T.; Uchida, H. AMPA receptors in schizophrenia: A systematic review of postmortem studies on receptor subunit expression and binding. Schizophr. Res. 2022, 243, 98–109. [Google Scholar] [CrossRef] [PubMed]
- Volk, D.W.; Eggan, S.M.; Lewis, D.A. Alterations in Metabotropic Glutamate Receptor 1α and Regulator of G Protein Signaling 4 in the Prefrontal Cortex in Schizophrenia. Am. J. Psychiatry 2010, 167, 1489–1498. [Google Scholar] [CrossRef] [Green Version]
- Ghose, S.; Crook, J.M.; Bartus, C.L.; Sherman, T.G.; Herman, M.M.; Hyde, T.M.; Kleinman, J.E.; Akil, M. Metabotropic Glutamate Receptor 2 and 3 Gene Expression in The Human Prefrontal Cortex and Mesencephalon in Schizophrenia. Int. J. Neurosci. 2008, 118, 1609–1627. [Google Scholar] [CrossRef] [Green Version]
- Bauer, D.; Gupta, D.; Harotunian, V.; Meador-Woodruff, J.H.; McCullumsmith, R.E. Abnormal expression of glutamate transporter and transporter interacting molecules in prefrontal cortex in elderly patients with schizophrenia. Schizophr. Res. 2008, 104, 108–120. [Google Scholar] [CrossRef] [Green Version]
- Parkin, G.M.; Gibbons, A.; Udawela, M.; Dean, B. Excitatory amino acid transporter (EAAT)1 and EAAT2 mRNA levels are altered in the prefrontal cortex of subjects with schizophrenia. J. Psychiatr. Res. 2020, 123, 151–158. [Google Scholar] [CrossRef]
- Scarr, E.; Udawela, M.; Dean, B. Changed frontal pole gene expression suggest altered interplay between neurotransmitter, developmental, and inflammatory pathways in schizophrenia. Schizophrenia 2018, 4, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauriat, T.; Dracheva, S.; Chin, B.; Schmeidler, J.; McInnes, L.; Haroutunian, V. Quantitative analysis of glutamate transporter mRNA expression in prefrontal and primary visual cortex in normal and schizophrenic brain. Neuroscience 2006, 137, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Hoftman, G.D.; Volk, D.W.; Bazmi, H.H.; Li, S.; Sampson, A.R.; Lewis, D.A. Altered Cortical Expression of GABA-Related Genes in Schizophrenia: Illness Progression vs Developmental Disturbance. Schizophr. Bull. 2013, 41, 180–191. [Google Scholar] [CrossRef]
- Torrey, E.F.; Barci, B.M.; Webster, M.J.; Bartko, J.J.; Meador-Woodruff, J.H.; Knable, M.B. Neurochemical markers for schizophrenia, bipolar disorder, and major depression in postmortem brains. Biol. Psychiatry 2005, 57, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Volk, D.W.; Austin, M.C.; Pierri, J.N.; Sampson, A.R.; Lewis, D.A. GABA Transporter-1 mRNA in the Prefrontal Cortex in Schizophrenia: Decreased Expression in a Subset of Neurons. Am. J. Psychiatry 2001, 158, 256–265. [Google Scholar] [CrossRef]
- Beneyto, M.; Abbott, A.; Hashimoto, T.; Lewis, D.A. Lamina-specific alterations in cortical GABA(A) receptor subunit expression in schizophrenia. Cereb. Cortex 2011, 21, 999–1011. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Lipska, B.K.; Halim, N.; Ma, Q.D.; Matsumoto, M.; Melhem, S.; Kolachana, B.S.; Hyde, T.M.; Herman, M.M.; Apud, J.; et al. Functional Analysis of Genetic Variation in Catechol-O-Methyltransferase (COMT): Effects on mRNA, Protein, and Enzyme Activity in Postmortem Human Brain. Am. J. Hum. Genet. 2004, 75, 807–821. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, M.; Weickert, C.S.; Beltaifa, S.; Kolachana, B.; Chen, J.; Hyde, T.M.; Herman, M.M.; Weinberger, D.R.; E Kleinman, J. Catechol O-Methyltransferase (COMT) mRNA Expression in the Dorsolateral Prefrontal Cortex of Patients with Schizophrenia. Neuropsychopharmacology 2003, 28, 1521–1530. [Google Scholar] [CrossRef] [Green Version]
- Kaalund, S.S.; Newburn, E.N.; Ye, T.; Tao, R.; Li, C.; A Deep-Soboslay, A.; Herman, M.M.; Hyde, T.M.; Weinberger, D.R.; Lipska, B.K.; et al. Contrasting changes in DRD1 and DRD2 splice variant expression in schizophrenia and affective disorders, and associations with SNPs in postmortem brain. Mol. Psychiatry 2013, 19, 1258–1266. [Google Scholar] [CrossRef]
- Stenkrona, P.; Matheson, G.J.; Halldin, C.; Cervenka, S.; Farde, L. D1-Dopamine Receptor Availability in First-Episode Neuroleptic Naive Psychosis Patients. Int. J. Neuropsychopharmacol. 2019, 22, 415–425. [Google Scholar] [CrossRef]
- Urigüen, L.; García-Fuster, M.J.; Callado, L.F.; Morentin, B.; La Harpe, R.; Casadó, V.; Lluis, C.; Franco, R.; García-Sevilla, J.A.; Meana, J.J. Immunodensity and mRNA expression of A2A adenosine, D2 dopamine, and CB1 cannabinoid receptors in postmortem frontal cortex of subjects with schizophrenia: Effect of antipsychotic treatment. Psychopharmacology 2009, 206, 313–324. [Google Scholar] [CrossRef]
- Meador-Woodruff, J.H.; Haroutunian, V.; Powchik, P.; Davidson, M.; Davis, K.L.; Watson, S.J. Dopamine Receptor Transcript Expression in Striatum and Prefrontal and Occipital Cortex. Arch. Gen. Psychiatry 1997, 54, 1089–1095. [Google Scholar] [CrossRef]
- Bray, N.J.; Preece, A.; Williams, N.M.; Moskvina, V.; Buckland, P.R.; Owen, M.J.; O’Donovan, M.C. Haplotypes at the dystrobrevin binding protein 1 (DTNBP1) gene locus mediate risk for schizophrenia through reduced DTNBP1 expression. Hum. Mol. Genet. 2005, 14, 1947–1954. [Google Scholar] [CrossRef] [Green Version]
- Baracskay, K.L.; Haroutunian, V.; Meador-Woodruff, J.H. Dopamine receptor signaling molecules are altered in elderly schizophrenic cortex. Synapse 2006, 60, 271–279. [Google Scholar] [CrossRef] [Green Version]
- Santarelli, D.M.; Carroll, A.P.; Cairns, H.M.; Tooney, P.A.; Cairns, M.J. Schizophrenia-associated MicroRNA-Gene Interactions in the Dorsolateral Prefrontal Cortex. Genom. Proteom. Bioinform. 2019, 17, 623–634. [Google Scholar] [CrossRef]
- Fung, S.J.; Sivagnanasundaram, S.; Weickert, C.S. Lack of Change in Markers of Presynaptic Terminal Abundance Alongside Subtle Reductions in Markers of Presynaptic Terminal Plasticity in Prefrontal Cortex of Schizophrenia Patients. Biol. Psychiatry 2011, 69, 71–79. [Google Scholar] [CrossRef] [Green Version]
- Hino, M.; Kunii, Y.; Matsumoto, J.; Wada, A.; Nagaoka, A.; Niwa, S.-I.; Takahashi, H.; Kakita, A.; Akatsu, H.; Hashizume, Y.; et al. Decreased VEGFR2 expression and increased phosphorylated Akt1 in the prefrontal cortex of individuals with schizophrenia. J. Psychiatr. Res. 2016, 82, 100–108. [Google Scholar] [CrossRef]
- Pillai, A. Decreased Expression of Sprouty2 in the Dorsolateral Prefrontal Cortex in Schizophrenia and Bipolar Disorder: A Correlation with BDNF Expression. PLoS ONE 2008, 3, e1784. [Google Scholar] [CrossRef] [Green Version]
- Iwamoto, K.; Bundo, M.; Yamada, K.; Takao, H.; Iwayama-Shigeno, Y.; Yoshikawa, T.; Kato, T. DNA Methylation Status of SOX10 Correlates with Its Downregulation and Oligodendrocyte Dysfunction in Schizophrenia. J. Neurosci. 2005, 25, 5376–5381. [Google Scholar] [CrossRef] [Green Version]
- Hakak, Y.; Walker, J.R.; Li, C.; Wong, W.H.; Davis, K.L.; Buxbaum, J.D.; Haroutunian, V.; Fienberg, A.A. Genome-wide expression analysis reveals dysregulation of myelination-related genes in chronic schizophrenia. Proc. Natl. Acad. Sci. USA 2001, 98, 4746–4751. [Google Scholar] [CrossRef]
- Santarelli, D.M.; Beveridge, N.J.; Tooney, P.A.; Cairns, M.J. Upregulation of Dicer and MicroRNA Expression in the Dorsolateral Prefrontal Cortex Brodmann Area 46 in Schizophrenia. Biol. Psychiatry 2011, 69, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Middleton, F.; Mirnics, K.; Pierri, J.N.; Lewis, D.; Levitt, P. Gene Expression Profiling Reveals Alterations of Specific Metabolic Pathways in Schizophrenia. J. Neurosci. 2002, 22, 2718–2729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arion, D.; Corradi, J.; Tang, S.; Datta, D.; Boothe, F.; He, A.; Cacace, A.M.; Zaczek, R.; Albright, C.F.; Tseng, G.; et al. Distinctive transcriptome alterations of prefrontal pyramidal neurons in schizophrenia and schizoaffective disorder. Mol. Psychiatry 2015, 20, 1397–1405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Verwer, R.W.; Lucassen, P.J.; Huitinga, I.; Swaab, D.F. Prefrontal cortex alterations in glia gene expression in schizophrenia with and without suicide. J. Psychiatr. Res. 2019, 121, 31–38. [Google Scholar] [CrossRef]
- Volk, D.W.; Moroco, A.E.; Roman, K.M.; Edelson, J.R.; Lewis, D.A. The Role of the Nuclear Factor-kappaB Transcriptional Complex in Cortical Immune Activation in Schizophrenia. Biol. Psychiatry 2019, 85, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Jo, Y.; Webster, M.J.; Lee, D. Shared co-expression networks in frontal cortex of the normal aged brain and schizophrenia. Schizophr. Res. 2018, 204, 253–261. [Google Scholar] [CrossRef]
- López-González, I.; Pinacho, R.; Vila, .; Escanilla, A.; Ferrer, I.; Ramos, B. Neuroinflammation in the dorsolateral prefrontal cortex in elderly chronic schizophrenia. Eur. Neuropsychopharmacol. 2019, 29, 384–396. [Google Scholar] [CrossRef] [Green Version]
- Hilker, R.; Helenius, D.; Fagerlund, B.; Skytthe, A.; Christensen, K.; Werge, T.M.; Nordentoft, M.; Glenthøj, B. Heritability of Schizophrenia and Schizophrenia Spectrum Based on the Nationwide Danish Twin Register. Biol. Psychiatry 2017, 83, 492–498. [Google Scholar] [CrossRef] [Green Version]
- Imamura, A.; Morimoto, Y.; Ono, S.; Kurotaki, N.; Kanegae, S.; Yamamoto, N.; Kinoshita, H.; Tsujita, T.; Okazaki, Y.; Ozawa, H. Genetic and environmental factors of schizophrenia and autism spectrum disorder: Insights from twin studies. J. Neural Transm. 2020, 127, 1501–1515. [Google Scholar] [CrossRef] [Green Version]
- Rajarajan, P.; Jiang, Y.; Kassim, B.S.; Akbarian, S. Chromosomal Conformations and Epigenomic Regulation in Schizophrenia. Prog. Mol. Biol. Transl. Sci. 2018, 157, 21–40. [Google Scholar]
- Micale, V.; Di Bartolomeo, M.; Di Martino, S.; Stark, T.; Dell’Osso, B.; Drago, F.; D’Addario, C. Are the epigenetic changes predictive of therapeutic efficacy for psychiatric disorders? A translational approach towards novel drug targets. Pharmacol. Ther. 2023, 241, 108279. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, M.V.C.; Bourc’His, D. The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell Biol. 2019, 20, 590–607. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.; Mendizabal, I.; Berto, S.; Chatterjee, P.; Layman, T.; Usui, N.; Toriumi, K.; Douglas, C.; Singh, D.; Huh, I.; et al. Evolution of DNA methylation in the human brain. Nat. Commun. 2021, 12, 2021. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, K.; Bundo, M.; Ueda, J.; Oldham, M.C.; Ukai, W.; Hashimoto, E.; Saito, T.; Geschwind, D.H.; Kato, T. Neurons show distinctive DNA methylation profile and higher interindividual variations compared with non-neurons. Genome Res. 2011, 21, 688–696. [Google Scholar] [CrossRef] [Green Version]
- Kozlenkov, A.; Wang, M.; Roussos, P.; Rudchenko, S.; Barbu, M.; Bibikova, M.; Klotzle, B.; Dwork, A.J.; Zhang, B.; Hurd, Y.L.; et al. Substantial DNA methylation differences between two major neuronal subtypes in human brain. Nucleic Acids Res. 2015, 44, 2593–2612. [Google Scholar] [CrossRef]
- E Jaffe, A.; Gao, Y.; Deep-Soboslay, A.; Tao, R.; Hyde, T.M.; Weinberger, D.R.; E Kleinman, J. Mapping DNA methylation across development, genotype and schizophrenia in the human frontal cortex. Nat. Neurosci. 2015, 19, 40–47. [Google Scholar] [CrossRef] [Green Version]
- Grayson, D.R.; Jia, X.; Chen, Y.; Sharma, R.P.; Mitchell, C.P.; Guidotti, A.; Costa, E. Reelin promoter hypermethylation in schizophrenia. Proc. Natl. Acad. Sci. USA 2005, 102, 9341–9346. [Google Scholar] [CrossRef] [Green Version]
- Mill, J.; Tang, T.; Kaminsky, Z.; Khare, T.; Yazdanpanah, S.; Bouchard, L.; Jia, P.; Assadzadeh, A.; Flanagan, J.; Schumacher, A.; et al. Epigenomic Profiling Reveals DNA-Methylation Changes Associated with Major Psychosis. Am. J. Hum. Genet. 2008, 82, 696–711. [Google Scholar] [CrossRef] [Green Version]
- Numata, S.; Ye, T.; Herman, M.; Lipska, B.K. DNA methylation changes in the postmortem dorsolateral prefrontal cortex of patients with schizophrenia. Front. Genet. 2014, 5, 280. [Google Scholar] [CrossRef] [Green Version]
- Tao, R.; Davis, K.N.; Li, C.; Shin, J.H.; Gao, Y.; Jaffe, A.; Gondré-Lewis, M.C.; Weinberger, D.R.; Kleinman, J.E.; Hyde, T.M. GAD1 alternative transcripts and DNA methylation in human prefrontal cortex and hippocampus in brain development, schizophrenia. Mol. Psychiatry 2017, 23, 1496–1505. [Google Scholar] [CrossRef]
- A Fachim, H.; Srisawat, U.; Dalton, C.F.; Reynolds, G.P. Parvalbumin promoter hypermethylation in postmortem brain in schizophrenia. Epigenomics 2018, 10, 519–524. [Google Scholar] [CrossRef] [PubMed]
- Tao, R.; Li, C.; Jaffe, A.E.; Shin, J.H.; Deep-Soboslay, A.; Yamin, R.; Weinberger, D.R.; Hyde, T.M.; Kleinman, J.E. Cannabinoid receptor CNR1 expression and DNA methylation in human prefrontal cortex, hippocampus and caudate in brain development and schizophrenia. Transl. Psychiatry 2020, 10, 158. [Google Scholar] [CrossRef]
- Wockner, L.; Noble, E.P.; Lawford, B.R.; Young, R.; Morris, P.; Whitehall, V.L.J.; Voisey, J. Genome-wide DNA methylation analysis of human brain tissue from schizophrenia patients. Transl. Psychiatry 2014, 4, e339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, D.; Chen, J.; Duan, K.; Perrone-Bizzozero, N.; Sui, J.; Calhoun, V.; Liu, J. Network modules linking expression and methylation in prefrontal cortex of schizophrenia. Epigenetics 2021, 16, 876–893. [Google Scholar] [CrossRef] [PubMed]
- Alelú-Paz, R.; Carmona, F.J.; Sanchez-Mut, J.V.; Cariaga-Martínez, A.; González-Corpas, A.; Ashour, N.; Orea, M.J.; Escanilla, A.; Monje, A.; Márquez, C.G.; et al. Epigenetics in Schizophrenia: A Pilot Study of Global DNA Methylation in Different Brain Regions Associated with Higher Cognitive Functions. Front. Psychol. 2016, 7, 1496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.; Lee, K.; Kim, K.; Yi, S.-J. The role of histone modifications: From neurodevelopment to neurodiseases. Signal Transduct. Target. Ther. 2022, 7, 217. [Google Scholar] [CrossRef] [PubMed]
- Girdhar, K.; Hoffman, G.E.; Jiang, Y.; Brown, L.; Kundakovic, M.; Hauberg, M.E.; Francoeur, N.J.; Wang, Y.-C.; Shah, H.; Kavanagh, D.H.; et al. Cell-specific histone modification maps in the human frontal lobe link schizophrenia risk to the neuronal epigenome. Nat. Neurosci. 2018, 21, 1126–1136. [Google Scholar] [CrossRef]
- Huang, H.-S.; Matevossian, A.; Whittle, C.; Kim, S.Y.; Schumacher, A.; Baker, S.P.; Akbarian, S. Prefrontal Dysfunction in Schizophrenia Involves Mixed-Lineage Leukemia 1-Regulated Histone Methylation at GABAergic Gene Promoters. J. Neurosci. 2007, 27, 11254–11262. [Google Scholar] [CrossRef] [Green Version]
- Gusev, F.E.; Reshetov, D.A.; Mitchell, A.C.; Andreeva, T.V.; Dincer, A.; Grigorenko, A.P.; Fedonin, G.; Halene, T.; Aliseychik, M.; Filippova, E.; et al. Chromatin profiling of cortical neurons identifies individual epigenetic signatures in schizophrenia. Transl. Psychiatry 2019, 9, 256. [Google Scholar] [CrossRef] [Green Version]
- Girdhar, K.; Hoffman, G.E.; Bendl, J.; Rahman, S.; Dong, P.; Liao, W.; Hauberg, M.E.; Sloofman, L.; Brown, L.; Devillers, O.; et al. Chromatin domain alterations linked to 3D genome organization in a large cohort of schizophrenia and bipolar disorder brains. Nat. Neurosci. 2022, 25, 474–483. [Google Scholar] [CrossRef]
- Nguyen, P.; Bar-Sela, G.; Sun, L.; Bisht, K.S.; Cui, H.; Kohn, E.; Feinberg, A.P.; Gius, D. BAT3 and SET1A Form a Complex with CTCFL/BORIS To Modulate H3K4 Histone Dimethylation and Gene Expression. Mol. Cell. Biol. 2008, 28, 6720–6729. [Google Scholar] [CrossRef] [PubMed]
- Singh, T.; I Kurki, M.; Curtis, D.; Purcell, S.M.; Crooks, L.; McRae, J.; Suvisaari, J.; Chheda, H.; Blackwood, D.; Breen, G.; et al. Rare loss-of-function variants in SETD1A are associated with schizophrenia and developmental disorders. Nat. Neurosci. 2016, 19, 571–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akbarian, S.; Ruehl, M.G.; Bliven, E.; Luiz, L.A.; Peranelli, A.C.; Baker, S.P.; Roberts, R.C.; Bunney, W.E.; Conley, R.C.; Jones, E.G.; et al. Chromatin Alterations Associated With Down-regulated Metabolic Gene Expression in the Prefrontal Cortex of Subjects With Schizophrenia. Arch. Gen. Psychiatry 2005, 62, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Dean, B.; A Thomas, E. Disease- and age-related changes in histone acetylation at gene promoters in psychiatric disorders. Transl. Psychiatry 2011, 1, e64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrelly, L.A.; Zheng, S.; Schrode, N.; Topol, A.; Bhanu, N.V.; Bastle, R.M.; Ramakrishnan, A.; Chan, J.C.; Cetin, B.; Flaherty, E.; et al. Chromatin profiling in human neurons reveals aberrant roles for histone acetylation and BET family proteins in schizophrenia. Nat. Commun. 2022, 13, 2195. [Google Scholar] [CrossRef]
- Sharma, R.P.; Grayson, D.R.; Gavin, D.P. Histone deactylase 1 expression is increased in the prefrontal cortex of schizophrenia subjects: Analysis of the National Brain Databank microarray collection. Schizophr. Res. 2008, 98, 111–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroeder, F.A.; Gilbert, T.M.; Feng, N.; Taillon, B.D.; Volkow, N.D.; Innis, R.B.; Hooker, J.M.; Lipska, B.K. Expression of HDAC2 but Not HDAC1 Transcript Is Reduced in Dorsolateral Prefrontal Cortex of Patients with Schizophrenia. ACS Chem. Neurosci. 2016, 8, 662–668. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, T.; Zürcher, N.R.; Wu, C.J.; Bhanot, A.; Hightower, B.G.; Kim, M.; Albrecht, D.S.; Wey, H.-Y.; Schroeder, F.A.; Rodriguez-Thompson, A.; et al. PET neuroimaging reveals histone deacetylase dysregulation in schizophrenia. J. Clin. Investig. 2018, 129, 364–372. [Google Scholar] [CrossRef] [Green Version]
- Esteller, M. Non-coding RNAs in human disease. Nat. Rev. Genet. 2011, 12, 861–874. [Google Scholar] [CrossRef]
- Cai, Y.; Yu, X.; Hu, S.; Yu, J. A Brief Review on the Mechanisms of miRNA Regulation. Genom. Proteom. Bioinform. 2009, 7, 147–154. [Google Scholar] [CrossRef] [Green Version]
- Morgunova, A.; Flores, C. MicroRNA regulation of prefrontal cortex development and psychiatric risk in adolescence. Semin. Cell Dev. Biol. 2021, 118, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Li, A.; Liu, Y.; Li, J.; Wang, M.; Sun, Y.; Qin, W.; Yu, C.; Jiang, T.; Liu, B. MIR137 polygenic risk is associated with schizophrenia and affects functional connectivity of the dorsolateral prefrontal cortex. Psychol. Med. 2019, 50, 1510–1518. [Google Scholar] [CrossRef] [PubMed]
- Beveridge, N.J.; Gardiner, E.; Carroll, A.P.; Tooney, P.; Cairns, M.J. Schizophrenia is associated with an increase in cortical microRNA biogenesis. Mol. Psychiatry 2009, 15, 1176–1189. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bilecki, W.; Maćkowiak, M. Gene Expression and Epigenetic Regulation in the Prefrontal Cortex of Schizophrenia. Genes 2023, 14, 243. https://doi.org/10.3390/genes14020243
Bilecki W, Maćkowiak M. Gene Expression and Epigenetic Regulation in the Prefrontal Cortex of Schizophrenia. Genes. 2023; 14(2):243. https://doi.org/10.3390/genes14020243
Chicago/Turabian StyleBilecki, Wiktor, and Marzena Maćkowiak. 2023. "Gene Expression and Epigenetic Regulation in the Prefrontal Cortex of Schizophrenia" Genes 14, no. 2: 243. https://doi.org/10.3390/genes14020243