
Dual Targeting of DNA Damage Response Proteins Implicated in Cancer Radioresistance
 , , and
, , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Structure-Based De Novo Dual Target Drug Design
2.1.1. Target Proteins
2.1.2. Ligand Building
2.1.3. Ligand Compound Naming
2.1.4. Drug-Likeness, Bioavailability, and Toxicity Evaluation
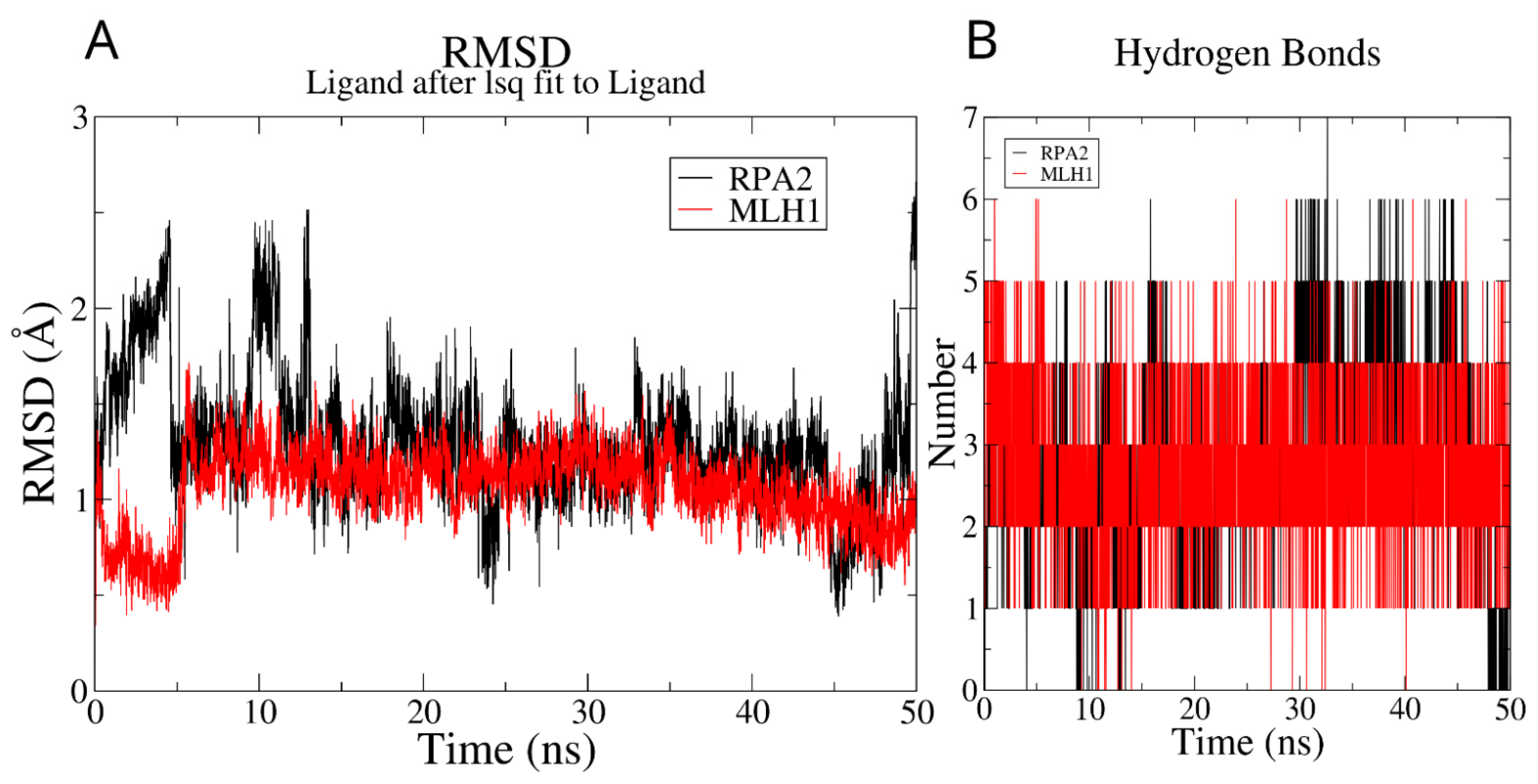
2.2. Molecular Dynamics Simulations
2.3. Calculation of Molecular Interaction Free Energies
2.4. Molecular Visualization
2.5. Virtual Screening of DDR/R proteins
2.5.1. Chemical Compound Selection and Structure Preparation
2.5.2. Pocket Identification and Analysis
2.5.3. Docking Experiments
3. Results
3.1. In Silico Drug Design
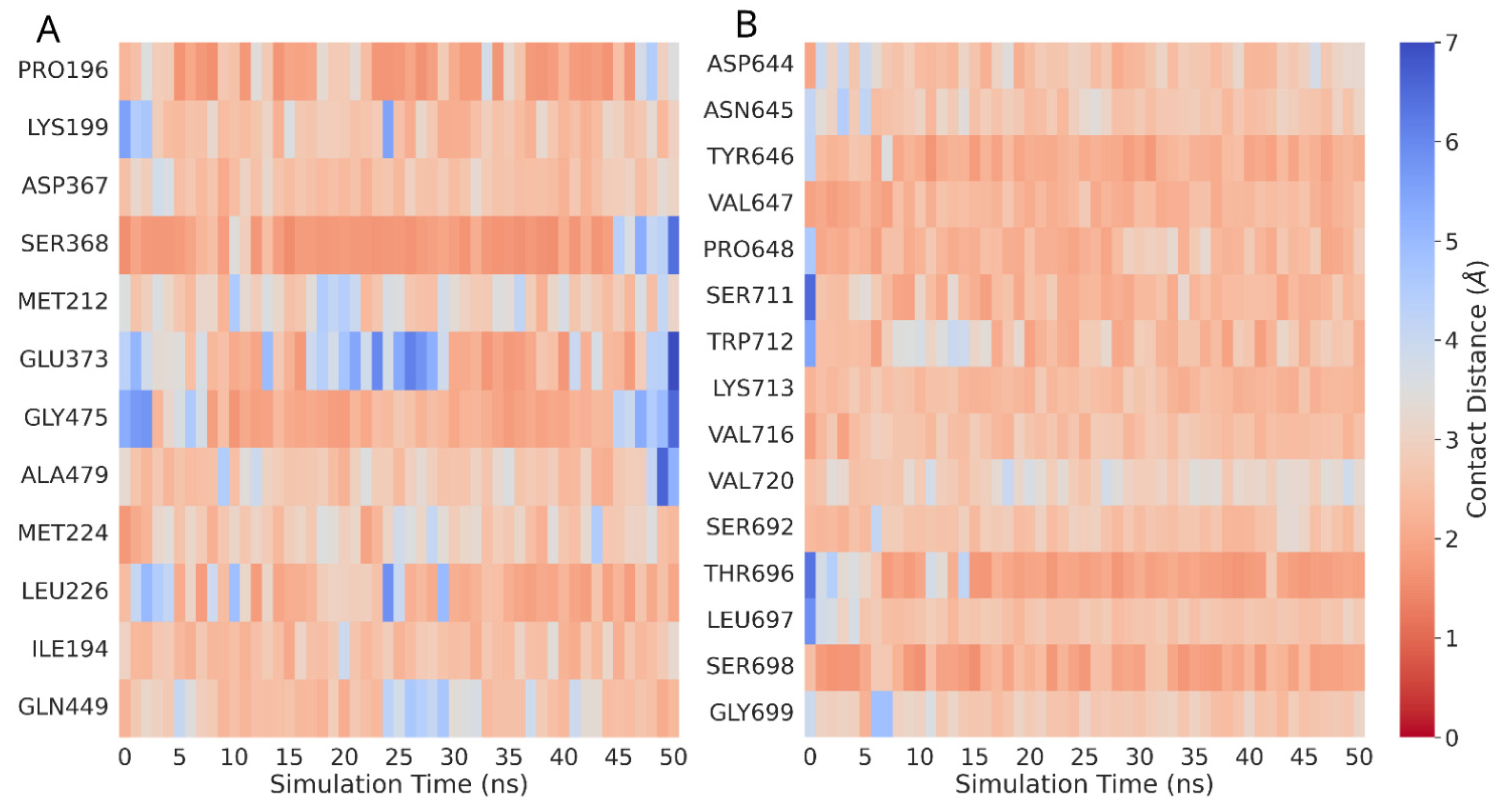
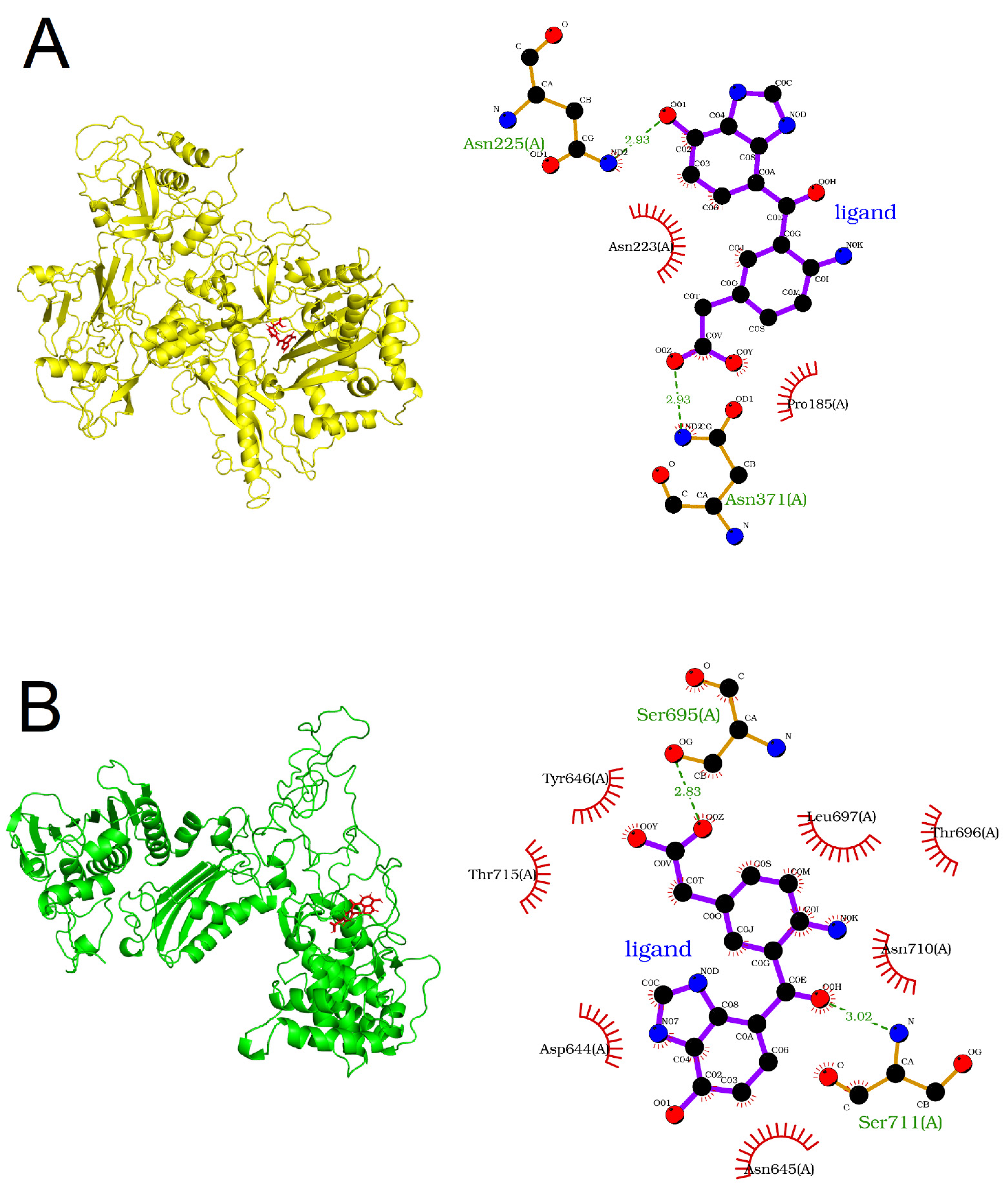
3.2. DDR Protein–Ligand Interaction Sites
3.3. Natural Compound Analogs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, J.J.; Lei, K.F.; Han, F. Tumor microenvironment: Recent advances in various cancer treatments. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 3855–3864. [Google Scholar]
- Baskar, R.; Lee, K.A.; Yeo, R.; Yeoh, K.W. Cancer and radiation therapy: Current advances and future directions. Int. J. Med. Sci. 2012, 9, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Toy, H.I.; Karakulah, G.; Kontou, P.I.; Alotaibi, H.; Georgakilas, A.G.; Pavlopoulou, A. Investigating molecular determinants of cancer cell resistance to ionizing radiation through an integrative bioinformatics approach. Front. Cell Dev. Biol. 2021, 9, 620248. [Google Scholar] [CrossRef]
- Chandra, R.A.; Keane, F.K.; Voncken, F.E.M.; Thomas, C.R., Jr. Contemporary radiotherapy: Present and future. Lancet 2021, 398, 171–184. [Google Scholar] [CrossRef] [PubMed]
- Borrego-Soto, G.; Ortiz-Lopez, R.; Rojas-Martinez, A. Ionizing radiation-induced DNA injury and damage detection in patients with breast cancer. Genet. Mol. Biol. 2015, 38, 420–432. [Google Scholar] [CrossRef] [PubMed]
- Malkova, A.; Haber, J.E. Mutations arising during repair of chromosome breaks. Ann. Rev. Genet. 2012, 46, 455–473. [Google Scholar] [CrossRef] [PubMed]
- Azzam, E.I.; Jay-Gerin, J.P.; Pain, D. Ionizing radiation-induced metabolic oxidative stress and prolonged cell injury. Cancer Lett. 2012, 327, 48–60. [Google Scholar] [CrossRef]
- Gong, L.; Zhang, Y.; Liu, C.; Zhang, M.; Han, S. Application of radiosensitizers in cancer radiotherapy. Int. J. Nanomed. 2021, 16, 1083–1102. [Google Scholar] [CrossRef]
- Liu, Y.P.; Zheng, C.C.; Huang, Y.N.; He, M.L.; Xu, W.W.; Li, B. Molecular mechanisms of chemo- and radiotherapy resistance and the potential implications for cancer treatment. MedComm 2021, 2, 315–340. [Google Scholar] [CrossRef]
- Nikitaki, Z.; Velalopoulou, A.; Zanni, V.; Tremi, I.; Havaki, S.; Kokkoris, M.; Gorgoulis, V.G.; Koumenis, C.; Georgakilas, A.G. Key biological mechanisms involved in high-let radiation therapies with a focus on DNA damage and repair. Expert Rev. Mol. Med. 2022, 24, e15. [Google Scholar] [CrossRef]
- Hada, M.; Georgakilas, A.G. Formation of clustered DNA damage after high-let irradiation: A review. J. Radiat. Res. 2008, 49, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Georgakilas, A.G.; O’Neill, P.; Stewart, R.D. Induction and repair of clustered DNA lesions: What do we know so far? Radiat. Res. 2013, 180, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Aziz, K.; Nowsheen, S.; Pantelias, G.; Iliakis, G.; Gorgoulis, V.G.; Georgakilas, A.G. Targeting DNA damage and repair: Embracing the pharmacological era for successful cancer therapy. Pharmacol. Ther. 2012, 133, 334–350. [Google Scholar] [CrossRef] [PubMed]
- Pavlopoulou, A.; Bagos, P.G.; Koutsandrea, V.; Georgakilas, A.G. Molecular determinants of radiosensitivity in normal and tumor tissue: A bioinformatic approach. Cancer Lett. 2017, 403, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Pavlopoulou, A.; Oktay, Y.; Vougas, K.; Louka, M.; Vorgias, C.E.; Georgakilas, A.G. Determinants of resistance to chemotherapy and ionizing radiation in breast cancer stem cells. Cancer Lett. 2016, 380, 485–493. [Google Scholar] [CrossRef]
- Wu, Y.; Song, Y.; Wang, R.; Wang, T. Molecular mechanisms of tumor resistance to radiotherapy. Mol. Cancer 2023, 22, 96. [Google Scholar] [CrossRef]
- Huang, R.X.; Zhou, P.K. DNA damage response signaling pathways and targets for radiotherapy sensitization in cancer. Signal Transduct. Target. Ther. 2020, 5, 60. [Google Scholar] [CrossRef]
- Hintelmann, K.; Kriegs, M.; Rothkamm, K.; Rieckmann, T. Improving the efficacy of tumor radiosensitization through combined molecular targeting. Front. Oncol. 2020, 10, 1260. [Google Scholar] [CrossRef]
- Maier, P.; Hartmann, L.; Wenz, F.; Herskind, C. Cellular pathways in response to ionizing radiation and their targetability for tumor radiosensitization. Int. J. Mol. Sci. 2016, 17, 102. [Google Scholar] [CrossRef]
- Qiu, Y.; Hu, X.; Zeng, X.; Wang, H. Triple kill: Ddr inhibitors, radiotherapy and immunotherapy leave cancer cells with no escape. Acta Biochim. Et Biophys. Sin. 2022, 54, 1569–1576. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Mirdita, M.; Schutze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. Colabfold: Making protein folding accessible to all. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Pei, J.; Lai, L. Ligbuilder v3: A multi-target de novo drug design approach. Front. Chem. 2020, 8, 142. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. Swissadme: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Pu, L.; Naderi, M.; Liu, T.; Wu, H.C.; Mukhopadhyay, S.; Brylinski, M. Etoxpred: A machine learning-based approach to estimate the toxicity of drug candidates. BMC Pharmacol. Toxicol. 2019, 20, 2. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.; Blundell, T.L.; Ascher, D.B. Pkcsm: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. Ucsf chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Crystal structure and pair potentials: A molecular-dynamics study. Phys. Rev. Lett. 1980, 45, 1196–1199. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef]
- Homeyer, N.; Gohlke, H. Free energy calculations by the molecular mechanics poisson-boltzmann surface area method. Mol. Inform. 2012, 31, 114–122. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. Ligplot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef] [PubMed]
- Sorokina, M.; Merseburger, P.; Rajan, K.; Yirik, M.A.; Steinbeck, C. Coconut online: Collection of open natural products database. J. Cheminform. 2021, 13, 2. [Google Scholar] [CrossRef] [PubMed]
- Le Guilloux, V.; Schmidtke, P.; Tuffery, P. Fpocket: An open source platform for ligand pocket detection. BMC Bioinform. 2009, 10, 168. [Google Scholar] [CrossRef] [PubMed]
- Koes, D.R.; Baumgartner, M.P.; Camacho, C.J. Lessons learned in empirical scoring with smina from the csar 2011 benchmarking exercise. J. Chem. Inf. Model. 2013, 53, 1893–1904. [Google Scholar] [CrossRef]
- Available online: https://www.cancer.gov/news-events/press-releases/2020 (accessed on 7 June 2021).
- Di Tommaso, P.; Chatzou, M.; Floden, E.W.; Barja, P.P.; Palumbo, E.; Notredame, C. Nextflow enables reproducible computational workflows. Nat. Biotechnol. 2017, 35, 316–319. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Pei, J.; Lai, L. Ligbuilder 2: A practical de novo drug design approach. J. Chem. Inf. Model. 2011, 51, 1083–1091. [Google Scholar] [CrossRef] [PubMed]
- Chagas, C.M.; Moss, S.; Alisaraie, L. Drug metabolites and their effects on the development of adverse reactions: Revisiting lipinski’s rule of five. Int. J. Pharm. 2018, 549, 133–149. [Google Scholar] [CrossRef]
- Martin, Y.C. A bioavailability score. J. Med. Chem. 2005, 48, 3164–3170. [Google Scholar] [CrossRef]
- Ertl, P.; Schuffenhauer, A. Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions. J. Cheminform. 2009, 1, 8. [Google Scholar] [CrossRef]
- Mortelmans, K.; Zeiger, E. The ames salmonella/microsome mutagenicity assay. Mutat. Res. 2000, 455, 29–60. [Google Scholar] [CrossRef]
- Liu, R.; Tawa, G.; Wallqvist, A. Locally weighted learning methods for predicting dose-dependent toxicity with application to the human maximum recommended daily dose. Chem. Res. Toxicol. 2012, 25, 2216–2226. [Google Scholar] [CrossRef] [PubMed]
- Hollingsworth, S.A.; Dror, R.O. Molecular dynamics simulation for all. Neuron 2018, 99, 1129–1143. [Google Scholar] [CrossRef] [PubMed]
- Paysan-Lafosse, T.; Blum, M.; Chuguransky, S.; Grego, T.; Pinto, B.L.; Salazar, G.A.; Bileschi, M.L.; Bork, P.; Bridge, A.; Colwell, L.; et al. Interpro in 2022. Nucleic Acids Res. 2023, 51, D418–D427. [Google Scholar] [CrossRef] [PubMed]
- Cramer, P.; Bushnell, D.A.; Kornberg, R.D. Structural basis of transcription: RNA polymerase ii at 2.8 angstrom resolution. Science 2001, 292, 1863–1876. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Kui, L.; Tang, M.; Li, D.; Wei, K.; Chen, W.; Miao, J.; Dong, Y. High-throughput transcriptome profiling in drug and biomarker discovery. Front. Genet. 2020, 11, 19. [Google Scholar] [CrossRef] [PubMed]
- Nikitaki, Z.; Michalopoulos, I.; Georgakilas, A.G. Molecular inhibitors of DNA repair: Searching for the ultimate tumor killing weapon. Future Med. Chem. 2015, 7, 1543–1558. [Google Scholar] [CrossRef]
- Raghavendra, N.M.; Pingili, D.; Kadasi, S.; Mettu, A.; Prasad, S. Dual or multi-targeting inhibitors: The next generation anticancer agents. Eur. J. Med. Chem. 2018, 143, 1277–1300. [Google Scholar] [CrossRef]
- Shi, D.; Khan, F.; Abagyan, R. Extended multitarget pharmacology of anticancer drugs. J. Chem. Inf. Model. 2019, 59, 3006–3017. [Google Scholar] [CrossRef]
- Taghipour, Y.D.; Zarebkohan, A.; Salehi, R.; Rahimi, F.; Torchilin, V.P.; Hamblin, M.R.; Seifalian, A. An update on dual targeting strategy for cancer treatment. J. Control. Release Off. J. Control. Release Soc. 2022, 349, 67–96. [Google Scholar] [CrossRef]
- Sleeth, K.M.; Sorensen, C.S.; Issaeva, N.; Dziegielewski, J.; Bartek, J.; Helleday, T. Rpa mediates recombination repair during replication stress and is displaced from DNA by checkpoint signalling in human cells. J. Mol. Biol. 2007, 373, 38–47. [Google Scholar] [CrossRef]
- Wold, M.S. Replication protein a: A heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annu. Rev. Biochem. 1997, 66, 61–92. [Google Scholar] [CrossRef] [PubMed]
- Dueva, R.; Iliakis, G. Replication protein a: A multifunctional protein with roles in DNA replication, repair and beyond. NAR Cancer 2020, 2, zcaa022. [Google Scholar] [CrossRef] [PubMed]
- Bendahhou, S.; Thoiron, B.; Duclohier, H. [Purification of sodium channel from squid mantle and reincorporation in planar lipid bilayers]. Comptes Rendus Acad. Sci. Ser. III Sci. Vie 1991, 312, 277–284. [Google Scholar]
- Block, W.D.; Yu, Y.; Lees-Miller, S.P. Phosphatidyl inositol 3-kinase-like serine/threonine protein kinases (pikks) are required for DNA damage-induced phosphorylation of the 32 kda subunit of replication protein a at threonine 21. Nucleic Acids Res. 2004, 32, 997–1005. [Google Scholar] [CrossRef] [PubMed]
- Anantha, R.W.; Sokolova, E.; Borowiec, J.A. Rpa phosphorylation facilitates mitotic exit in response to mitotic DNA damage. Proc. Natl. Acad. Sci. USA 2008, 105, 12903–12908. [Google Scholar] [CrossRef]
- Sancar, A.; Lindsey-Boltz, L.A.; Unsal-Kacmaz, K.; Linn, S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 2004, 73, 39–85. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Liu, Y.; Wu, X.; Shell, S.M. Functions of human replication protein a (rpa): From DNA replication to DNA damage and stress responses. J. Cell. Physiol. 2006, 208, 267–273. [Google Scholar] [CrossRef]
- Golub, E.I.; Gupta, R.C.; Haaf, T.; Wold, M.S.; Radding, C.M. Interaction of human rad51 recombination protein with single-stranded DNA binding protein, rpa. Nucleic Acids Res. 1998, 26, 5388–5393. [Google Scholar] [CrossRef]
- Nimonkar, A.V.; Genschel, J.; Kinoshita, E.; Polaczek, P.; Campbell, J.L.; Wyman, C.; Modrich, P.; Kowalczykowski, S.C. Blm-dna2-rpa-mrn and exo1-blm-rpa-mrn constitute two DNA end resection machineries for human DNA break repair. Genes Dev. 2011, 25, 350–362. [Google Scholar] [CrossRef]
- Zhao, S.; Xu, B.; Ma, W.; Chen, H.; Jiang, C.; Cai, J.; Meng, X. DNA damage repair in brain tumor immunotherapy. Front. Immunol. 2021, 12, 829268. [Google Scholar] [CrossRef]
- Pedersen, H.; Anne Adanma Obara, E.; Elbaek, K.J.; Vitting-Serup, K.; Hamerlik, P. Replication protein a (rpa) mediates radio-resistance of glioblastoma cancer stem-like cells. Int. J. Mol. Sci. 2020, 21, 1588. [Google Scholar] [CrossRef] [PubMed]
- Morita, R.; Nakane, S.; Shimada, A.; Inoue, M.; Iino, H.; Wakamatsu, T.; Fukui, K.; Nakagawa, N.; Masui, R.; Kuramitsu, S. Molecular mechanisms of the whole DNA repair system: A comparison of bacterial and eukaryotic systems. J. Nucleic Acids 2010, 2010, 179594. [Google Scholar] [CrossRef] [PubMed]
- Dahai, Y.; Sanyuan, S.; Hong, L.; Di, Z.; Chong, Z. A relationship between replication protein a and occurrence and prognosis of esophageal carcinoma. Cell Biochem. Biophys. 2013, 67, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Fourtziala, E.; Givalos, N.; Alexakis, N.; Griniatsos, J.; Alevizopoulos, N.; Kavantzas, N.; Lazaris, A.C.; Korkolopoulou, P.; Gakiopoulou, H. Replication protein a (rpa1, rpa2 and rpa3) expression in gastric cancer: Correlation with clinicopathologic parameters and patients’ survival. Off. J. Balk. Union Oncol. 2020, 25, 1482–1489. [Google Scholar]
- Algethami, M.; Toss, M.S.; Woodcock, C.L.; Jaipal, C.; Brownlie, J.; Shoqafi, A.; Alblihy, A.; Mesquita, K.A.; Green, A.R.; Mongan, N.P.; et al. Unravelling the clinicopathological and functional significance of replication protein a (rpa) heterotrimeric complex in breast cancers. NPJ Breast Cancer 2023, 9, 18. [Google Scholar] [CrossRef] [PubMed]
- Di, Z.; Sanyuan, S.; Hong, L.; Dahai, Y. Enhanced radiosensitivity and g2/m arrest were observed in radioresistant esophageal cancer cells by knocking down rpa expression. Cell Biochem. Biophys. 2014, 70, 887–891. [Google Scholar] [CrossRef] [PubMed]
- Glanzer, J.G.; Liu, S.; Oakley, G.G. Small molecule inhibitor of the rpa70 n-terminal protein interaction domain discovered using in silico and in vitro methods. Bioorganic Med. Chem. 2011, 19, 2589–2595. [Google Scholar] [CrossRef]
- Glanzer, J.G.; Carnes, K.A.; Soto, P.; Liu, S.; Parkhurst, L.J.; Oakley, G.G. A small molecule directly inhibits the p53 transactivation domain from binding to replication protein a. Nucleic Acids Res. 2013, 41, 2047–2059. [Google Scholar] [CrossRef]
- Glanzer, J.G.; Liu, S.; Wang, L.; Mosel, A.; Peng, A.; Oakley, G.G. Rpa inhibition increases replication stress and suppresses tumor growth. Cancer Res. 2014, 74, 5165–5172. [Google Scholar] [CrossRef]
- Shuck, S.C.; Turchi, J.J. Targeted inhibition of replication protein a reveals cytotoxic activity, synergy with chemotherapeutic DNA-damaging agents, and insight into cellular function. Cancer Res. 2010, 70, 3189–3198. [Google Scholar] [CrossRef]
- Dueva, R.; Krieger, L.M.; Li, F.; Luo, D.; Xiao, H.; Stuschke, M.; Metzen, E.; Iliakis, G. Chemical inhibition of rpa by hamno alters cell cycle dynamics by impeding DNA replication and g2-to-m transition but has little effect on the radiation-induced DNA damage response. Int. J. Mol. Sci. 2023, 24, 14941. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Jiang, Y.; Liu, J.; Liu, J.; Shi, M.; Chen, J.; Zhang, J.; Tian, Y.; Yang, X.; Liu, H. Targeting rpa promotes autophagic flux and the antitumor response to radiation in nasopharyngeal carcinoma. J. Transl. Med. 2023, 21, 738. [Google Scholar] [CrossRef] [PubMed]
- Fishel, R. Mismatch repair. J. Biol. Chem. 2015, 290, 26395–26403. [Google Scholar] [CrossRef]
- Chahwan, R.; van Oers, J.M.; Avdievich, E.; Zhao, C.; Edelmann, W.; Scharff, M.D.; Roa, S. The atpase activity of mlh1 is required to orchestrate DNA double-strand breaks and end processing during class switch recombination. J. Exp. Med. 2012, 209, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Fishel, R.; Lescoe, M.K.; Rao, M.R.; Copeland, N.G.; Jenkins, N.A.; Garber, J.; Kane, M.; Kolodner, R. The human mutator gene homolog msh2 and its association with hereditary nonpolyposis colon cancer. Cell 1993, 75, 1027–1038. [Google Scholar] [CrossRef] [PubMed]
- Ellison, A.R.; Lofing, J.; Bitter, G.A. Human mutl homolog (mlh1) function in DNA mismatch repair: A prospective screen for missense mutations in the atpase domain. Nucleic Acids Res. 2004, 32, 5321–5338. [Google Scholar] [CrossRef] [PubMed]
- Lynch, H.T.; Snyder, C.L.; Shaw, T.G.; Heinen, C.D.; Hitchins, M.P. Milestones of lynch syndrome: 1895–2015. Nat. Rev. Cancer 2015, 15, 181–194. [Google Scholar] [CrossRef]
- Peltomaki, P. Epigenetic mechanisms in the pathogenesis of lynch syndrome. Clin. Genet. 2014, 85, 403–412. [Google Scholar] [CrossRef]
- Reyes, G.X.; Schmidt, T.T.; Kolodner, R.D.; Hombauer, H. New insights into the mechanism of DNA mismatch repair. Chromosoma 2015, 124, 443–462. [Google Scholar] [CrossRef]
- Lee, V.; Murphy, A.; Le, D.T.; Diaz, L.A., Jr. Mismatch repair deficiency and response to immune checkpoint blockade. Oncologist 2016, 21, 1200–1211. [Google Scholar] [CrossRef]
- Huang, Y.; Feng, L.; Bao, Y.; Zhang, Y.; Liang, J.; Mao, Q.; Li, J.; Jiang, C. Expressing mlh1 in hct116 cells increases cellular resistance to radiation by activating the prkac. Exp. Biol. Med. 2022, 247, 426–432. [Google Scholar] [CrossRef] [PubMed]
- Holt, S.M.; Scemama, J.L.; Panayiotidis, M.I.; Georgakilas, A.G. Compromised repair of clustered DNA damage in the human acute lymphoblastic leukemia msh2-deficient nalm-6 cells. Mutat. Res. 2009, 674, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Pilie, P.G.; Tang, C.; Mills, G.B.; Yap, T.A. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 81–104. [Google Scholar] [CrossRef]
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M.; International Natural Product Sciences, T.; Supuran, C.T. Natural products in drug discovery: Advances and opportunities. Nat. Rev. Drug Discov. 2021, 20, 200–216. [Google Scholar] [CrossRef] [PubMed]
- Theofylaktou, D.; Takan, I.; Karakulah, G.; Biz, G.M.; Zanni, V.; Pavlopoulou, A.; Georgakilas, A.G. Mining natural products with anticancer biological activity through a systems biology approach. Oxidative Med. Cell. Longev. 2021, 2021, 9993518. [Google Scholar] [CrossRef] [PubMed]
- Topatana, W.; Juengpanich, S.; Li, S.; Cao, J.; Hu, J.; Lee, J.; Suliyanto, K.; Ma, D.; Zhang, B.; Chen, M.; et al. Advances in synthetic lethality for cancer therapy: Cellular mechanism and clinical translation. J. Hematol. Oncol. 2020, 13, 118. [Google Scholar] [CrossRef] [PubMed]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in brca mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Patterson-Fortin, J.; Bose, A.; Tsai, W.C.; Grochala, C.; Nguyen, H.; Zhou, J.; Parmar, K.; Lazaro, J.B.; Liu, J.; McQueen, K.; et al. Targeting DNA repair with combined inhibition of nhej and mmej induces synthetic lethality in tp53-mutant cancers. Cancer Res. 2022, 82, 3815–3829. [Google Scholar] [CrossRef]
- Deneka, A.Y.; Einarson, M.B.; Bennett, J.; Nikonova, A.S.; Elmekawy, M.; Zhou, Y.; Lee, J.W.; Burtness, B.A.; Golemis, E.A. Synthetic lethal targeting of mitotic checkpoints in hpv-negative head and neck cancer. Cancers 2020, 12, 306. [Google Scholar] [CrossRef]
- Morgan, M.A.; Lawrence, T.S. Molecular pathways: Overcoming radiation resistance by targeting DNA damage response pathways. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 2898–2904. [Google Scholar] [CrossRef]
- Knijnenburg, T.A.; Wang, L.; Zimmermann, M.T.; Chambwe, N.; Gao, G.F.; Cherniack, A.D.; Fan, H.; Shen, H.; Way, G.P.; Greene, C.S.; et al. Genomic and molecular landscape of DNA damage repair deficiency across the cancer genome atlas. Cell Rep. 2018, 23, 239–254.e236. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Huang, J.; Liang, D.; Hu, Y.; Mao, B.; Li, Q.; Sun, H.; Yang, Y.; Zhang, J.; Zhang, H.; et al. DNA damage repair gene mutations are indicative of a favorable prognosis in colorectal cancer treated with immune checkpoint inhibitors. Front. Oncol. 2020, 10, 549777. [Google Scholar] [CrossRef] [PubMed]
- Colton, M.; Cheadle, E.J.; Honeychurch, J.; Illidge, T.M. Reprogramming the tumour microenvironment by radiotherapy: Implications for radiotherapy and immunotherapy combinations. Radiat. Oncol. 2020, 15, 254. [Google Scholar] [CrossRef] [PubMed]
- Lhuillier, C.; Rudqvist, N.P.; Elemento, O.; Formenti, S.C.; Demaria, S. Radiation therapy and anti-tumor immunity: Exposing immunogenic mutations to the immune system. Genome Med. 2019, 11, 40. [Google Scholar] [CrossRef]
- Zhao, P.; Li, L.; Jiang, X.; Li, Q. Mismatch repair deficiency/microsatellite instability-high as a predictor for anti-pd-1/pd-l1 immunotherapy efficacy. J. Hematol. Oncol. 2019, 12, 54. [Google Scholar] [CrossRef]
- Durante, M.; Formenti, S.C. Radiation-induced chromosomal aberrations and immunotherapy: Micronuclei, cytosolic DNA, and interferon-production pathway. Front. Oncol. 2018, 8, 192. [Google Scholar] [CrossRef]
- Spiotto, M.; Fu, Y.X.; Weichselbaum, R.R. The intersection of radiotherapy and immunotherapy: Mechanisms and clinical implications. Sci. Immunol. 2016, 1, eaag1266. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Energies (kJ/mol) | RPA2 + Ligand | MLH + Ligand |
|---|---|---|
| Van der Waal | −80.69 ± 2.27 | −185.46 ± 1.64 |
| Electrostatic | −64.74 ± 4.01 | −94.66 ± 2.31 |
| Polar solvation | 106.51 ± 3.30 | 122.22 ± 1.57 |
| SASA | −15.13 ± 0.25 | −17.58 ± 0.12 |
| Binding | −54.04 ± 2.77 | −175.48 ± 1.98 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vasilopoulos, S.N.; Güner, H.; Uça Apaydın, M.; Pavlopoulou, A.; Georgakilas, A.G. Dual Targeting of DNA Damage Response Proteins Implicated in Cancer Radioresistance. Genes 2023, 14, 2227. https://doi.org/10.3390/genes14122227
Vasilopoulos SN, Güner H, Uça Apaydın M, Pavlopoulou A, Georgakilas AG. Dual Targeting of DNA Damage Response Proteins Implicated in Cancer Radioresistance. Genes. 2023; 14(12):2227. https://doi.org/10.3390/genes14122227
Chicago/Turabian StyleVasilopoulos, Spyridon N., Hüseyin Güner, Merve Uça Apaydın, Athanasia Pavlopoulou, and Alexandros G. Georgakilas. 2023. "Dual Targeting of DNA Damage Response Proteins Implicated in Cancer Radioresistance" Genes 14, no. 12: 2227. https://doi.org/10.3390/genes14122227