
Cornelia de Lange Syndrome Caused by an Intragenic Heterozygous Deletion in RAD21 Detected through Very-High-Resolution Chromosomal Microarray Analysis

Abstract
:1. Introduction
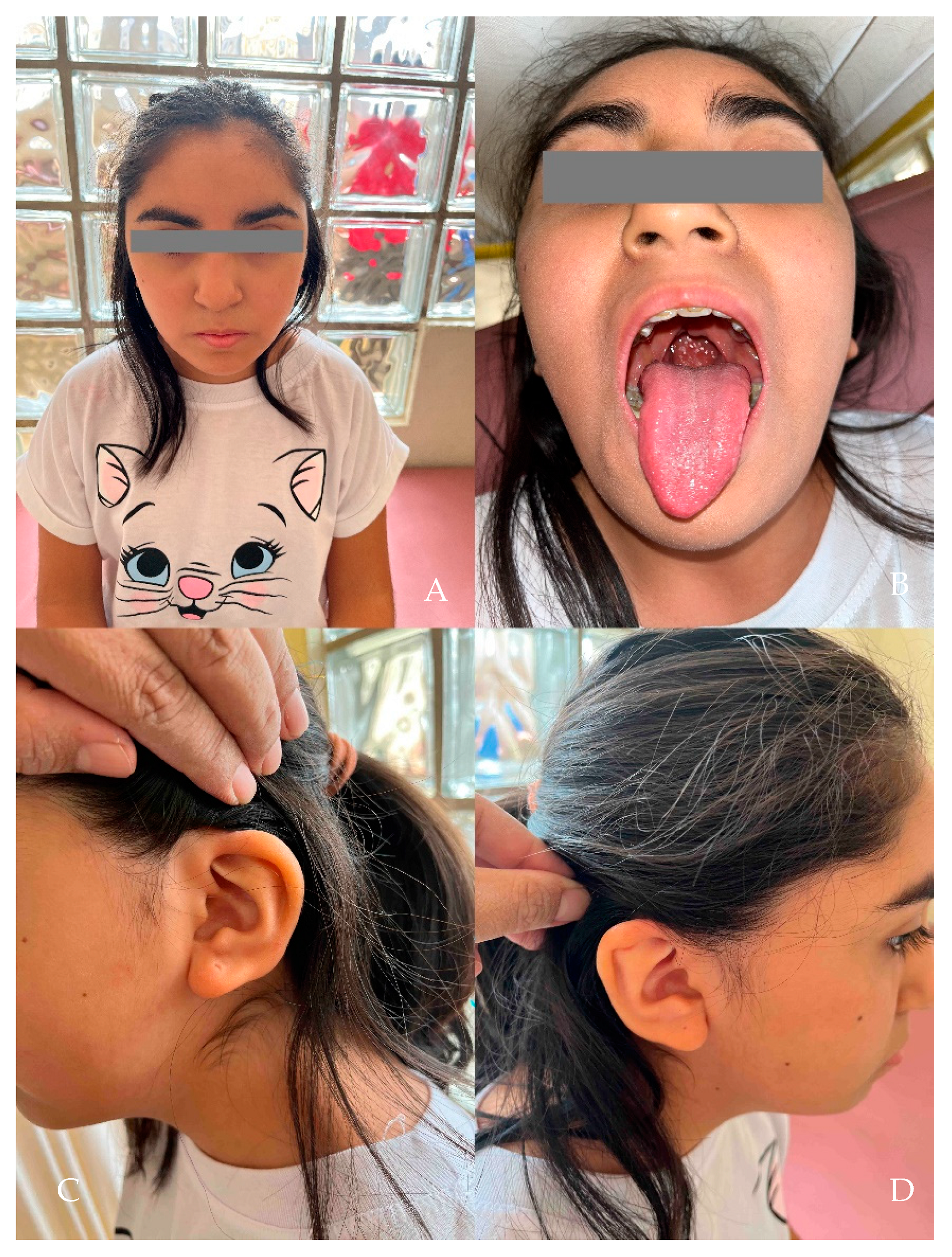
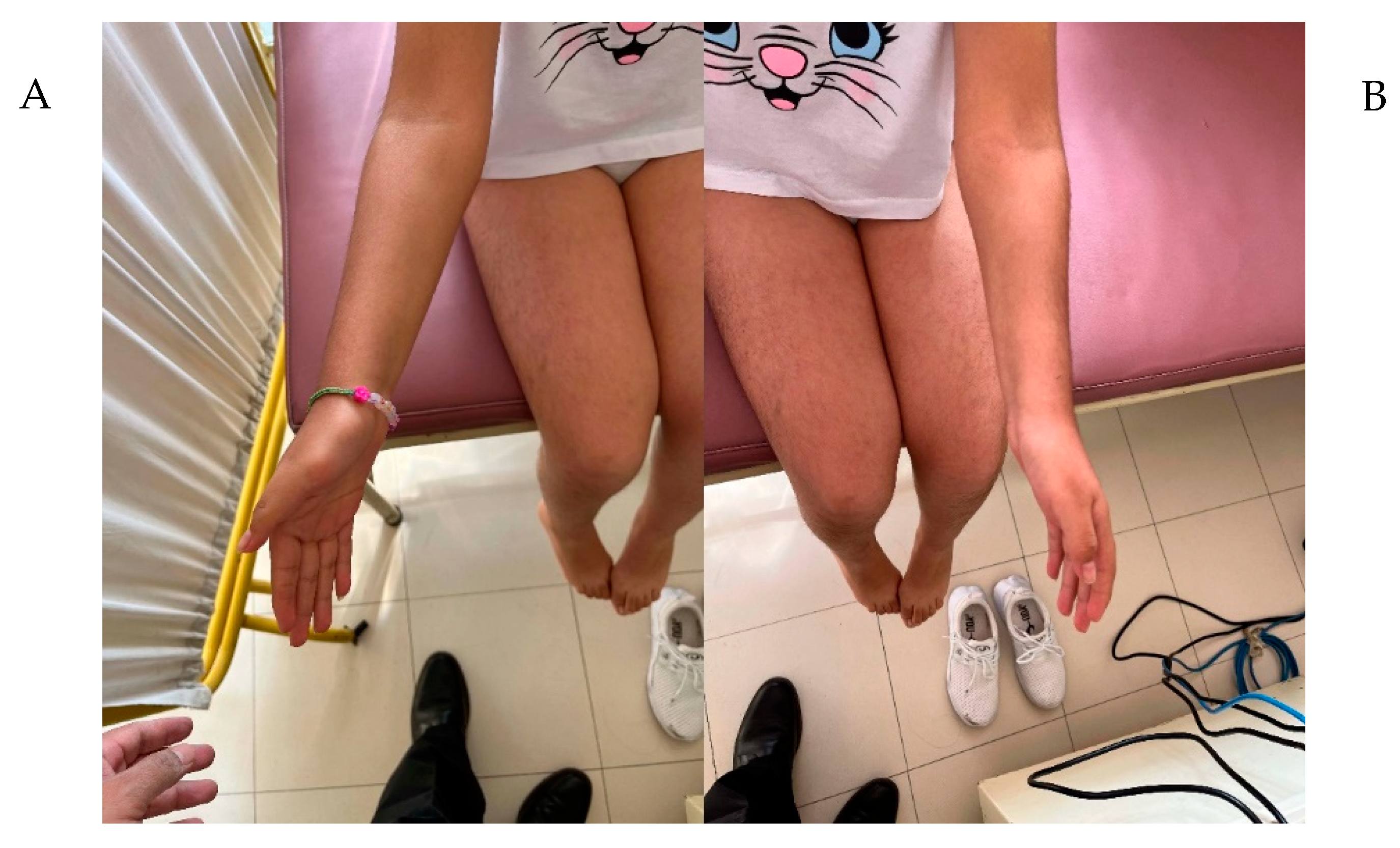
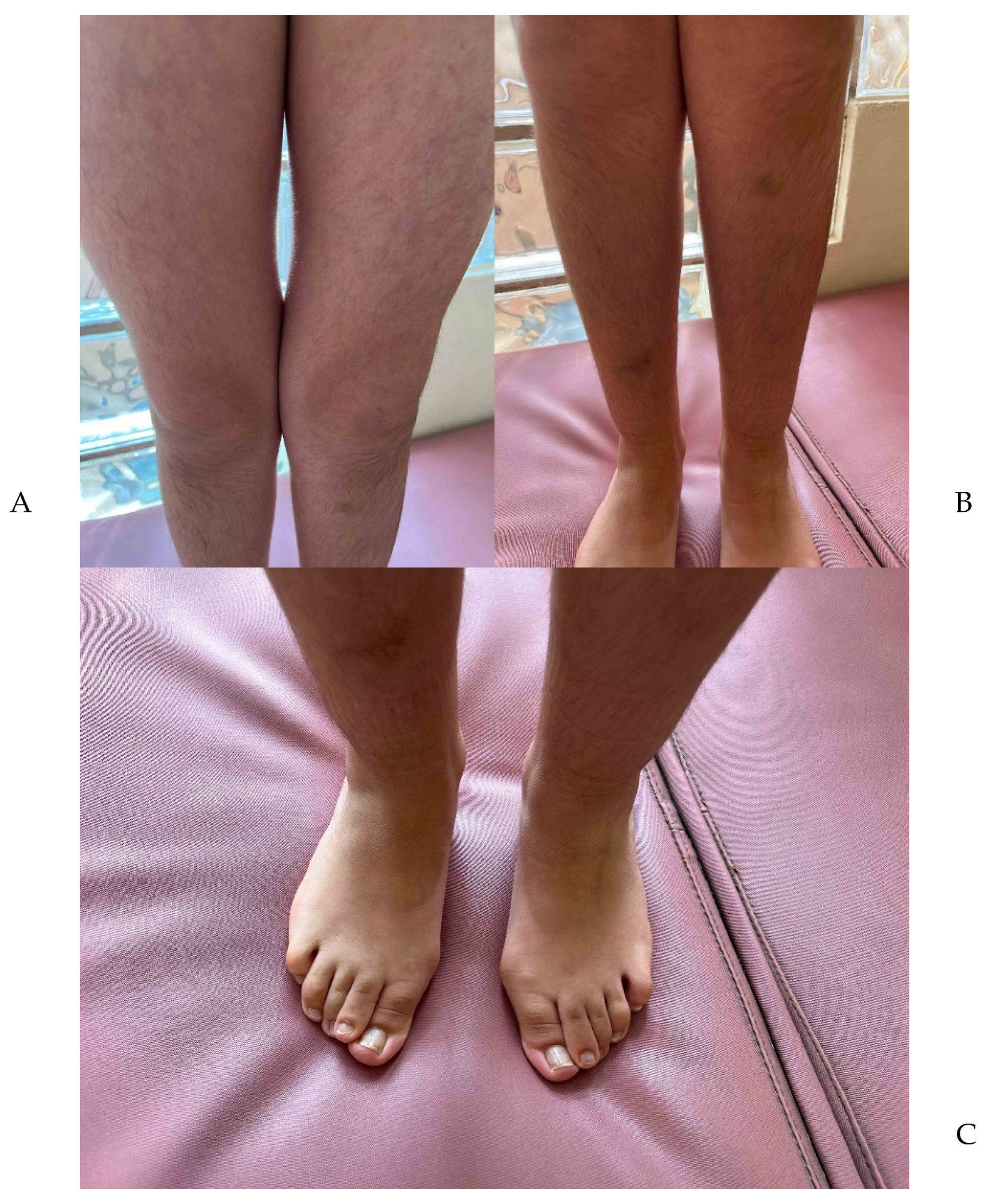
2. Results
Clinical Evaluation
3. Materials and Methods
3.1. Ethical Statement
3.2. Chromosome Microarray Analysis
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- OMIM—Online Mendelian Inheritance in Man. Available online: http://www.omim.org/ (accessed on 15 May 2016).
- Kline, A.D.; Moss, J.F.; Selicorni, A.; Bisgaard, A.-M.; Deardorff, M.A.; Gillett, P.M.; Ishman, S.L.; Kerr, L.M.; Levin, A.V.; Mulder, P.A.; et al. Diagnosis and management of Cornelia de Lange syndrome: First international consensus statement. Nat. Rev. Genet. 2018, 19, 649–666. [Google Scholar] [CrossRef] [PubMed]
- Parenti, I.; Kaiser, F.J. Cornelia de Lange Syndrome as Paradigm of Chromatinopathies. Front. Neurosci. 2021, 15, 774950. [Google Scholar] [CrossRef] [PubMed]
- Kline, A.D.; Krantz, I.D.; Sommer, A.; Kliewer, M.; Jackson, L.G.; FitzPatrick, D.R.; Levin, A.V.; Selicorni, A. Cornelia de Lange syndrome: Clinical review, diagnostic and scoring systems, and anticipatory guidance. Am. J. Med. Genet. A 2007, 143A, 1287–1296. [Google Scholar] [CrossRef] [PubMed]
- Deschamps, G.N. Cornelia de Lange Syndrome. Neonatal. Netw. 2022, 41, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Gudmundsson, S.; Annerén, G.; Marcos-Alcalde, Í.; Wilbe, M.; Melin, M.; Gómez-Puertas, P.; Bondeson, M.-L. A novel RAD21 p.(Gln592del) variant expands the clinical description of Cornelia de Lange syndrome type 4—Review of the literature. Eur. J. Med. Genet. 2019, 62, 103526. [Google Scholar] [CrossRef] [PubMed]
- Bonora, E.; Bianco, F.; Cordeddu, L.; Bamshad, M.; Francescatto, L.; Dowless, D.; Stanghellini, V.; Cogliandro, R.F.; Lindberg, G.; Mungan, Z.; et al. Mutations in RAD21 disrupt regulation of APOB in patients with chronic intestinal pseudo-obstruction. Gastroenterology 2015, 148, 771–782.e11. [Google Scholar] [CrossRef] [PubMed]
- Krab, L.C.; Marcos-Alcalde, I.; Assaf, M.; Balasubramanian, M.; Andersen, J.B.; Bisgaard, A.-M.; Fitzpatrick, D.R.; Gudmundsson, S.; Huisman, S.A.; Kalayci, T.; et al. Delineation of phenotypes and genotypes related to cohesin structural protein RAD21. Hum. Genet. 2020, 139, 575–592. [Google Scholar] [CrossRef]
- McKay, M.J.; Troelstra, C.; Kanaar, R.; Smit, B.; Hagemeijer, A.; Bootsma, D.; Hoeijmakers, J.H. Sequence conservation of the rad21 Schizosaccharomyces pombe DNA double-strand break repair gene in human and mouse. Genomics 1996, 36, 305–315. [Google Scholar] [CrossRef]
- Sumara, I.; Vorlaufer, E.; Gieffers, C.; Peters, B.H.; Peters, J.-M. Characterization of Vertebrate Cohesin Complexes and Their Regulation in Prophase. J. Cell Biol. 2000, 151, 749–762. [Google Scholar] [CrossRef]
- Cheng, H.; Zhang, N.; Pati, D. Cohesin subunit RAD21: From biology to disease. Gene 2020, 758, 144966. [Google Scholar] [CrossRef]
- Xu, H.; Balakrishnan, K.; Malaterre, J.; Beasley, M.; Yan, Y.; Essers, J.; Appeldoorn, E.; Thomaszewski, J.M.; Vazquez, M.; McKay, M.J.; et al. Rad21-cohesin haploinsufficiency impedes DNA repair and enhances gastrointestinal radiosensitivity in mice. PLoS ONE 2010, 5, e12112. [Google Scholar] [CrossRef]
- Chen, F.; Kamradt, M.; Mulcahy, M.; Byun, Y.; Xu, H.; McKay, M.J.; Cryns, V.L. Caspase proteolysis of the cohesin component RAD21 promotes apoptosis. J. Biol. Chem. 2002, 277, 16775–16781. [Google Scholar] [CrossRef] [PubMed]
- Pati, D.; Zhang, N.; Plon, S.E. Linking sister chromatid cohesion and apoptosis: Role of Rad21. Mol. Cell. Biol. 2002, 22, 8267–8277. [Google Scholar] [CrossRef] [PubMed]
- Beauchene, N.A.; Díaz-Martínez, L.A.; Furniss, K.; Hsu, W.-S.; Tsai, H.-J.; Chamberlain, C.; Esponda, P.; Giménez-Abián, J.F.; Clarke, D.J. Rad21 is required for centrosome integrity in human cells independently of its role in chromosome cohesion. Cell Cycle 2010, 9, 1774–1780. [Google Scholar] [CrossRef]
- Seitan, V.C.; Hao, B.; Tachibana-Konwalski, K.; Lavagnolli, T.; Mira-Bontenbal, H.; Brown, K.E.; Teng, G.; Carroll, T.; Terry, A.; Horan, K.; et al. A role for cohesin in T-cell-receptor rearrangement and thymocyte differentiation. Nature 2011, 476, 467–471. [Google Scholar] [CrossRef]
- Fisher, J.B.; Peterson, J.; Reimer, M.; Stelloh, C.; Pulakanti, K.; Gerbec, Z.J.; Abel, A.M.; Strouse, J.M.; Strouse, C.; McNulty, M.; et al. The cohesin subunit Rad21 is a negative regulator of hematopoietic self-renewal through epigenetic repression of Hoxa7 and Hoxa9. Leukemia 2017, 31, 712–719. [Google Scholar] [CrossRef]
- Zhang, Y.; da Fang, Y. Progresses on the structure and function of cohesin. Hereditas 2020, 42, 57–72. [Google Scholar]
- Deardorff, M.A.; Wilde, J.J.; Albrecht, M.; Dickinson, E.; Tennstedt, S.; Braunholz, D.; Mönnich, M.; Yan, Y.; Xu, W.; Gil-Rodríguez, M.C.; et al. RAD21 Mutations Cause a Human Cohesinopathy. Am. J. Hum. Genet. 2012, 90, 1014. [Google Scholar] [CrossRef]
- Ansari, M.; Poke, G.; Ferry, Q.; Williamson, K.; Aldridge, R.; Meynert, A.M.; Bengani, H.; Chan, C.Y.; Kayserili, H.; Avci, Ş.; et al. Genetic heterogeneity in Cornelia de Lange syndrome (CdLS) and CdLS-like phenotypes with observed and predicted levels of mosaicism. J. Med. Genet. 2014, 51, 659–668. [Google Scholar] [CrossRef]
- Minor, A.; Shinawi, M.; Hogue, J.S.; Vineyard, M.; Hamlin, D.R.; Tan, C.; Donato, K.; Wysinger, L.; Botes, S.; Das, S.; et al. Two novel RAD21 mutations in patients with mild Cornelia de Lange syndrome-like presentation and report of the first familial case. Gene 2014, 537, 279–284. [Google Scholar] [CrossRef]
- Lee, H.; Deignan, J.L.; Dorrani, N.; Strom, S.P.; Kantarci, S.; Quintero-Rivera, F.; Das, K.; Toy, T.; Harry, B.; Yourshaw, M.; et al. Clinical exome sequencing for genetic identification of rare Mendelian disorders. JAMA 2014, 312, 1880–1887. [Google Scholar] [CrossRef] [PubMed]
- Martínez, F.; Caro-Llopis, A.; Roselló, M.; Oltra, S.; Mayo, S.; Monfort, S.; Orellana, C. High diagnostic yield of syndromic intellectual disability by targeted next-generation sequencing. J. Med. Genet. 2017, 54, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Boyle, M.I.; Jespersgaard, C.; Nazaryan, L.; Ravn, K.; Brøndum-Nielsen, K.; Bisgaard, A.-M.; Tümer, Z. Deletion of 11q12.3–11q13.1 in a patient with intellectual disability and childhood facial features resembling Cornelia de Lange syndrome. Gene 2015, 572, 130–134. [Google Scholar] [CrossRef] [PubMed]
- Yuan, B.; Neira, J.; Pehlivan, D.; Santiago-Sim, T.; Song, X.; Rosenfeld, J.; Posey, J.E.; Patel, V.; Jin, W.; Adam, M.P. Clinical exome sequencing reveals locus heterogeneity and phenotypic variability of cohesinopathies. Genet. Med. Off. J. Am. Coll. Med. Genet. 2019, 21, 663–675. [Google Scholar] [CrossRef]
- Kruszka, P.; Berger, S.I.; Casa, V.; Dekker, M.R.; Gaesser, J.; Weiss, K.; Martinez, A.F.; Murdock, D.R.; Louie, R.J.; Prijoles, E.J.; et al. Cohesin complex-associated holoprosencephaly. Brain 2019, 142, 2631–2643. [Google Scholar] [CrossRef]
- Dorval, S.; Masciadri, M.; Mathot, M.; Russo, S.; Revencu, N.; Larizza, L. A novel RAD21 mutation in a boy with mild Cornelia de Lange presentation: Further delineation of the phenotype. Eur. J. Med. Genet. 2020, 63, 103620. [Google Scholar] [CrossRef]
- Latorre-Pellicer, A.; Ascaso, Á.; Trujillano, L.; Gil-Salvador, M.; Arnedo, M.; Lucia-Campos, C.; Antoñanzas-Pérez, R.; Marcos-Alcalde, I.; Parenti, I.; Bueno-Lozano, G.; et al. Evaluating Face2Gene as a Tool to Identify Cornelia de Lange Syndrome by Facial Phenotypes. Int. J. Mol. Sci. 2020, 21, 1042. [Google Scholar] [CrossRef]
- Lei, M.; Liang, D.; Yang, Y.; Mitsuhashi, S.; Katoh, K.; Miyake, N.; Frith, M.C.; Wu, L.; Matsumoto, N. Long-read DNA sequencing fully characterized chromothripsis in a patient with Langer-Giedion syndrome and Cornelia de Lange syndrome-4. J. Hum. Genet. 2020, 65, 667–674. [Google Scholar] [CrossRef]
- Abarca-Barriga, H.H.; Vásquez-Sotomayor, F. Utilidad diagnóstica de la secuenciación de segunda y tercera generación en pacientes con discapacidad intelectual: Revisión rápida. Psiquiatr. Biológica 2023, 30, 100392. [Google Scholar] [CrossRef]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Aguilera, M.A.; Meyer, R.; Massouras, A. VarSome: The human genomic variant search engine. Bioinformatics 2019, 35, 1978–1980. [Google Scholar] [CrossRef]
- Rodrigues, E.d.S.; Griffith, S.; Martin, R.; Antonescu, C.; Posey, J.E.; Coban-Akdemir, Z.; Jhangiani, S.N.; Doheny, K.F.; Lupski, J.R.; Valle, D.; et al. Variant-level matching for diagnosis and discovery: Challenges and opportunities. Hum. Mutat. 2022, 43, 782–790. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Deardorff et al. 2012 [19] (n = 6) | Minor et al. 2014 [21] (n = 2) | Ansari et al. 2014 [20] (n = 1) | Lee et al. 2014 [22] (n = 1) | Martínez et al. 2017 [23] (n = 1) | Boyle et al. 2017 [24] (n = 4) | Yuan et al. 2019 [25] (n = 2) | Kruszka et al. 2019 [26] (n = 3) | Gudmunson et al. 2019 [6] (n = 1) | Dorval et al. 2019 [27] (n = 1) | Latorre-Pellicer et al. 2020 [28] (n = 3) | Lei et al. 2020 [29] (n = 1) | Abarca et al. 2023 [30] (n = 1) | Total (N = 26) | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 1 | 2 | 1 | 2 | 3 | 4 | 1 | 2 | 1 | 2 | 3 | 1 | 2 | 3 | (%) | |||||||||
| Sex | * | M | * | * | * | * | M | M | F | * | * | F | F | F | F | * | * | F | M | M | M | M | F | F | M | M | F | F = 9/M = 9/* = 9 | |
| Age (years) | * | * | * | * | * | * | 3 | 12 | 4 | * | 26 | * | * | * | * | * | 7 | 14 | 2 | 1.25 | 5 | 3 | 5 | 8 | 6 | 13 | Mdn = 5.5 | ||
| Congenital microcephaly | * | + | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | − | * | * | * | * | + | 2/3 | 66.7 |
| Small for gestationatl age | * | + | * | * | * | * | + | + | * | * | * | * | * | * | * | * | * | * | * | * | − | − | * | * | * | * | − | 3/6 | 50 |
| Low weight | + | + | − | * | − | * | + | − | − | * | * | * | * | * | * | * | * | * | * | * | + | * | * | * | * | + | + | 4/10 | 60 |
| Short stature | + | + | − | + | − | * | + | − | − | * | * | * | * | * | * | * | * | − | − | − | − | * | * | * | * | + | + | 6/14 | 42.9 |
| Holoprosencephaly | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | + | + | − | * | − | * | * | * | * | − | 2/5 | 40 |
| Microcephaly | + | + | + | + | + | + | + | + | + | + | * | + | + | * | * | * | * | * | + | + | + | + | * | * | * | * | + | 17/17 | 100 |
| Prominent forehead | − | * | * | * | − | + | + | − | * | * | * | − | * | * | * | * | * | * | * | * | * | * | * | * | * | * | + | 3/7 | 42.9 |
| Braquicephaly | + | − | * | * | * | * | − | + | * | * | * | * | * | * | * | * | * | * | * | + | * | * | * | * | * | * | − | 3/6 | 50 |
| Thick eyebrow | + | + | + | + | + | + | + | + | + | + | * | + | * | * | * | * | * | * | + | + | + | + | * | * | * | + | + | 17/17 | 100 |
| Synophrys | + | − | + | + | + | + | + | + | * | * | * | * | * | * | * | * | * | + | * | + | − | + | * | * | * | * | − | 10/13 | 76.9 |
| Nistagmus | − | * | * | * | * | * | + | − | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | − | 1/4 | 25 |
| Hyperopia | − | * | * | * | * | * | − | + | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | − | 1/4 | 25 |
| Long eyelashes | + | + | + | + | + | + | − | + | * | + | * | * | * | * | * | * | * | * | * | * | + | + | * | * | * | + | + | 11/14 | 92.3 |
| Long philtrum | − | + | + | + | − | + | − | + | * | * | * | + | * | * | * | * | * | * | + | + | + | + | * | * | * | * | + | 11/14 | 78.6 |
| Strabismus | * | * | * | * | * | + | + | − | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | − | 2/4 | 50 |
| Ptosis | + | * | * | * | + | + | + | − | * | * | * | + | * | * | * | * | * | * | * | * | + | * | * | * | * | * | + | 7/8 | 87.5 |
| Micrognathia | + | − | + | + | − | − | + | − | * | * | * | − | * | * | * | * | * | * | * | * | + | * | * | * | * | + | + | 7/12 | 58.3 |
| Cleft palate | + | − | * | * | + | * | * | * | * | * | * | + | * | * | + | * | + | + | * | * | * | * | + | + | 8/9 | 88.9 | |||
| Broad uvula | * | * | * | * | * | * | − | + | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | − | 1/3 | 33.3 |
| Short nose | + | * | * | * | * | − | + | − | * | * | * | + | * | * | * | * | * | * | * | + | + | * | * | * | * | * | + | 6/8 | 75 |
| Nares anteverted | * | * | * | * | * | + | − | + | * | * | * | + | * | * | * | * | * | * | + | * | + | * | * | * | * | + | − | 6/8 | 75 |
| Depressed nasal bridge | + | + | * | * | * | − | + | − | * | * | * | + | * | * | * | * | * | * | * | * | * | * | * | * | * | + | − | 5/8 | 62.5 |
| High nasal bridge | * | * | * | * | * | + | − | + | * | * | * | − | * | * | * | * | * | * | * | * | * | * | * | * | * | * | + | 3/5 | 60 |
| Increased posterior angulation of ear | + | * | * | * | * | + | + | − | * | * | * | − | * | * | * | * | * | * | * | * | * | * | * | * | * | * | + | 4/6 | 66.7 |
| Low-set ear | * | * | * | * | * | * | − | + | * | * | * | − | * | * | * | * | * | * | + | + | * | * | * | * | * | * | − | 3/6 | 50 |
| Deafness | − | * | * | * | * | * | − | − | * | * | * | * | * | + | + | * | * | * | * | * | − | * | * | * | * | * | − | 2/7 | 28.6 |
| Hirsutism | − | − | * | + | * | − | − | + | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | + | 3/7 | 42.9 |
| Clinodactyly of fitth finger | + | * | * | − | * | + | + | + | * | * | * | * | * | * | * | * | * | * | + | + | + | + | * | * | * | + | − | 9/11 | 81.8 |
| Syndactily of 2-3 fingers | * | * | * | − | * | + | − | + | * | * | * | * | * | * | * | * | * | * | * | * | − | * | * | * | * | * | − | 2/6 | 33.3 |
| Overlapping toes (2-3) | * | * | * | − | * | * | + | − | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | + | 2/4 | 50 |
| Syndactily of 2-3 toes | * | * | * | − | + | + | + | − | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | − | 3/6 | 50 |
| Single transverse palmar crease | * | * | * | * | * | * | + | − | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | − | 1/3 | 33.3 |
| Radio-ulna synostosis, limited elbow range of motion | + | − | * | − | + | * | * | * | * | * | + | + | * | * | * | * | * | − | * | * | * | * | * | + | 5/8 | 62.5 | |||
| Hemivertebra/butterfly vertebra | + | + | * | * | + | * | * | * | * | * | * | * | * | * | * | − | − | * | * | * | * | * | + | − | 4/10 | 57.1 | |||
| Pectus excavatum | * | * | * | * | − | * | − | + | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | − | 1/4 | 25 |
| Pectus carinatum | * | * | * | * | + | * | − | − | − | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | − | 1/5 | 20 |
| Congenital heart disease | * | * | * | * | + | * | * | * | * | * | * | + | * | * | − | * | − | * | * | * | * | * | + | − | 3/6 | 50 | |||
| Gastroesophageal reflux disease | + | * | * | * | + | * | − | + | * | * | * | * | * | * | * | * | * | * | + | + | + | + | * | * | * | * | − | 7/9 | 77.8 |
| Hypospadias | * | * | * | * | * | * | + | − | n/a | * | * | n/a | n/a | n/a | n/a | * | * | n/a | * | * | * | * | n/a | n/a | * | * | n/a | 1/2 | 50 |
| Cryptorchidism | * | * | * | * | * | * | + | − | n/a | * | * | n/a | n/a | n/a | n/a | * | * | n/a | * | * | + | * | n/a | n/a | * | * | n/a | 2/3 | 66.7 |
| Bilateral inguinal hernia | * | * | * | * | * | * | + | − | n/a | * | * | n/a | n/a | n/a | n/a | * | * | n/a | * | * | * | * | n/a | n/a | * | + | n/a | 2/3 | 66.7 |
| Congenital diaphragmatic hernia | * | * | * | * | * | * | * | * | n/a | * | * | n/a | n/a | n/a | n/a | * | * | n/a | * | * | + | * | n/a | n/a | * | * | n/a | 1/1 | 100 |
| Bifid scrotum | * | * | * | * | * | * | + | − | n/a | * | * | n/a | n/a | n/a | n/a | * | * | n/a | * | * | * | * | n/a | n/a | * | * | n/a | 1/2 | 50 |
| Penoscrotal transposition | * | + | * | * | * | * | + | − | n/a | * | * | n/a | n/a | n/a | n/a | * | * | n/a | * | * | * | * | n/a | n/a | * | * | n/a | 2/3 | 66.7 |
| Development delay | − | * | + | * | + | * | + | + | * | * | * | * | * | * | * | * | * | + | + | + | + | − | * | * | * | * | − | 8/11 | 72.7 |
| Language delay | − | + | + | * | + | * | + | − | * | + | * | * | * | * | * | * | * | * | * | * | * | + | * | * | * | * | − | 6/9 | 66.7 |
| Motor delay | − | + | * | * | + | * | + | − | * | * | * | * | * | * | * | * | * | * | * | * | * | − | * | * | * | * | − | 3/7 | 42.9 |
| Autistic spectrum disorders | − | * | * | * | + | * | + | . | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | − | 2/4 | 50 |
| Specific learning disorders | − | * | * | * | * | * | − | + | * | * | * | + | + | + | + | * | * | * | * | * | * | * | * | * | * | * | − | 5/8 | 62.5 |
| Intellectual disability | − | * | + | + | * | * | * | * | * | * | * | * | * | * | * | * | * | − | − | * | * | * | + | − | 3/7 | 42.9 | |||
| ADHD | − | * | * | * | * | * | + | + | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | − | 2/4 | 50 |
| Cutis marmorata | + | + | * | − | * | − | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | + | 3/5 | 60 |
| Preaxial polydactyly | + | 1/1 | 100 | ||||||||||||||||||||||||||
| Bicornuate uterus | + | 1/1 | 100 | ||||||||||||||||||||||||||
| Clinical scores | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | − | 10 | * | * | ||
| Variant | 8:117,708,713–121,024,193 ❡ | 8:117,640,909–119,330,085 ❡ | 8:117237890–122631628 ❡ | 8:116,950,003–118,944,486 ❡ | c.1127C>G | c.1753T>C | Deletion exon 13 | c.592_593dup | * | c.1808T>C | c.86G>A | c.704delG | c.704delG | c.704delG | c.704delG | c.1550dupC | c.1161+1G>A | c.1548delinsTC | c.589C>T | c.1217_1224del | c.1774_1776del | c.943_946del | c.1382C>T | 8:117765326_122494596 ❡ | 8:117765326_118270323 ❡ | 8:115443000_123744000₣ | 8:116845458_116854956₣ | ||
| Exon | 1–14 | 1–14 | 1–14 | 1–14 | 9 | 14 | 13 | 3 | 14 | 2 | 7 | 7 | 7 | 7 | 12 | Intron 10 | 12 | 6 | 10 | 14 | 9 | 11 | 1–14 | 1–14 | 1–14 | 9–14 | |||
| 665 pb | * | ||||||||||||||||||||||||||||
| Type of variant | CNV | CNV | CNV | CNV | Missense | Missense | Frameshift | Frameshift | Splicing site | Missense | Codon stop | Codon stop | Codon stop | Codon stop | Codon stop | Frameshift | Splicing site | Frameshift | Codon stop | Frameshift | Frameshift | Codon stop | Missense | CNV | CNV | CNV | Frameshift | ||
| Classification | LP | LP | P | P | P | P | P | P | P | LP | P | VUS−LB | P | P | P | P | |||||||||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abarca-Barriga, H.H.; Punil Luciano, R.; Vásquez Sotomayor, F. Cornelia de Lange Syndrome Caused by an Intragenic Heterozygous Deletion in RAD21 Detected through Very-High-Resolution Chromosomal Microarray Analysis. Genes 2023, 14, 2212. https://doi.org/10.3390/genes14122212
Abarca-Barriga HH, Punil Luciano R, Vásquez Sotomayor F. Cornelia de Lange Syndrome Caused by an Intragenic Heterozygous Deletion in RAD21 Detected through Very-High-Resolution Chromosomal Microarray Analysis. Genes. 2023; 14(12):2212. https://doi.org/10.3390/genes14122212
Chicago/Turabian StyleAbarca-Barriga, Hugo H., Renzo Punil Luciano, and Flor Vásquez Sotomayor. 2023. "Cornelia de Lange Syndrome Caused by an Intragenic Heterozygous Deletion in RAD21 Detected through Very-High-Resolution Chromosomal Microarray Analysis" Genes 14, no. 12: 2212. https://doi.org/10.3390/genes14122212