
Population Structure and Genetic Diversity of Yunling Cattle Determined by Whole-Genome Resequencing

Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples and Sequencing
2.2. Assemble and Annotation
2.3. Reads Mapping and SNPs Calling
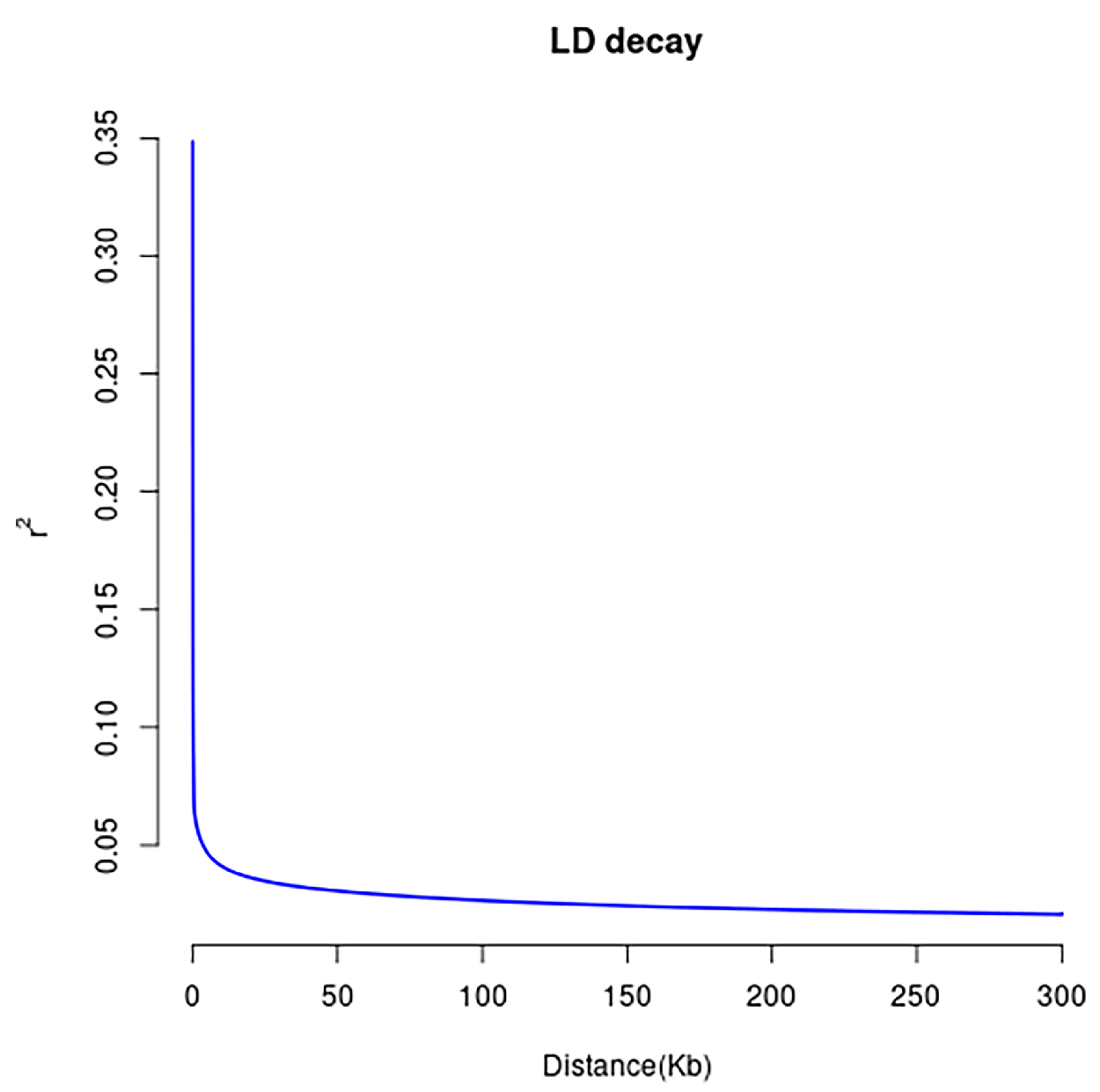
2.4. Linkage Disequilibrium and Genetic Diversity Analysis
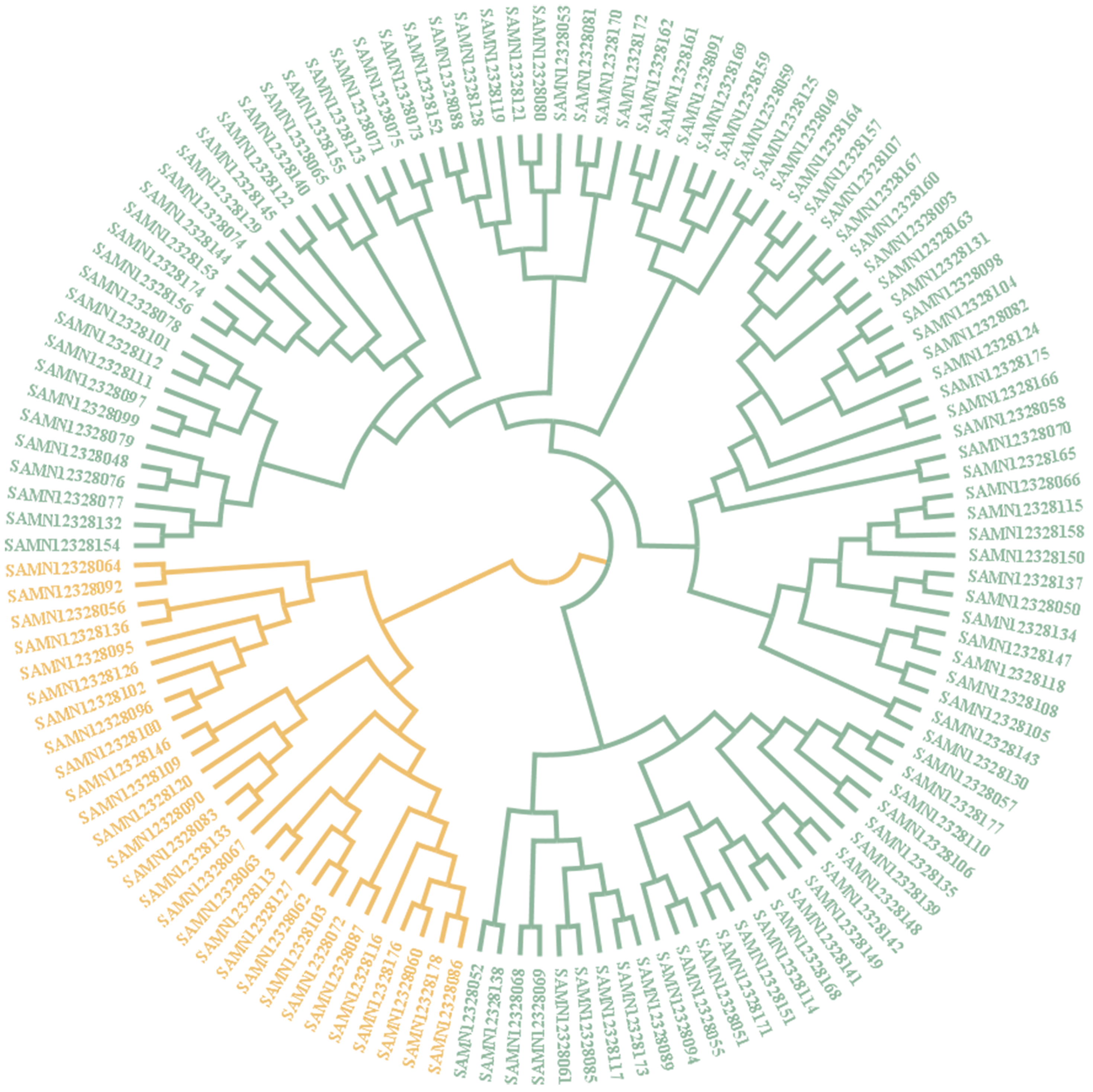
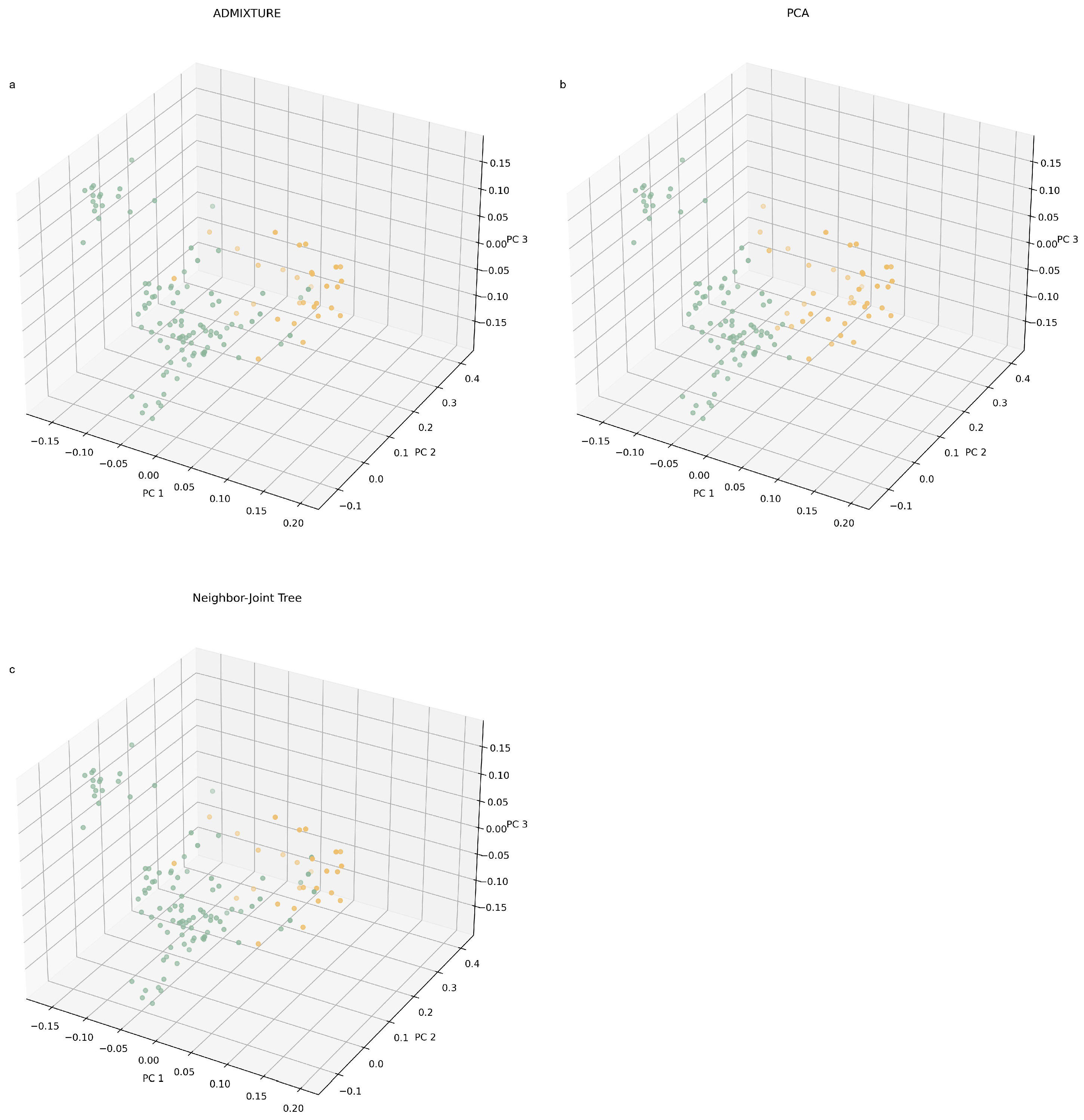
2.5. Population Structure, Principal Component Analysis and Phylogenetic Tree Analysis
2.6. Inbreeding Analysis and Subgroup Analysis
3. Results
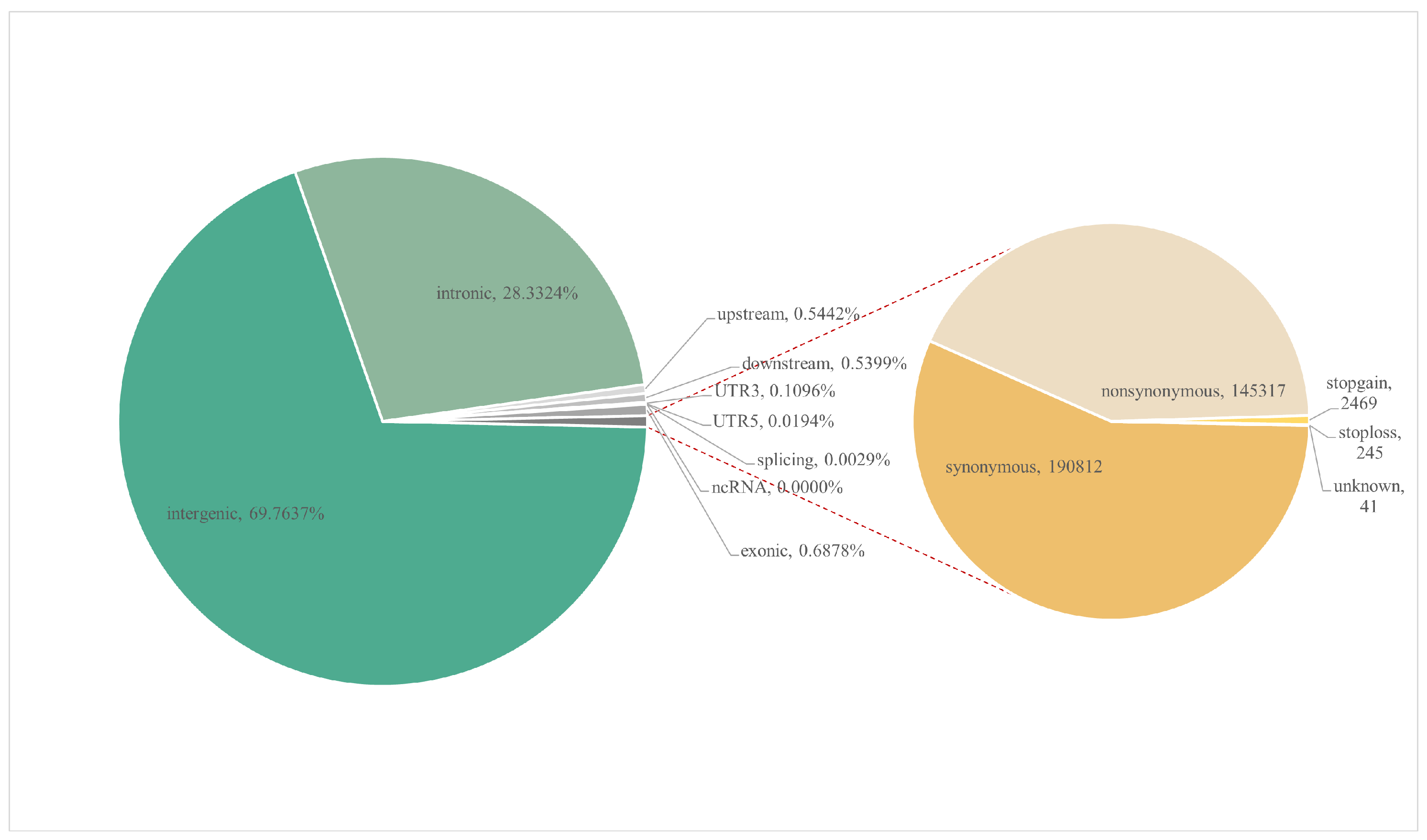
3.1. Diversity in Single-Nucleotide Polymorphisms, Genetic Diversity
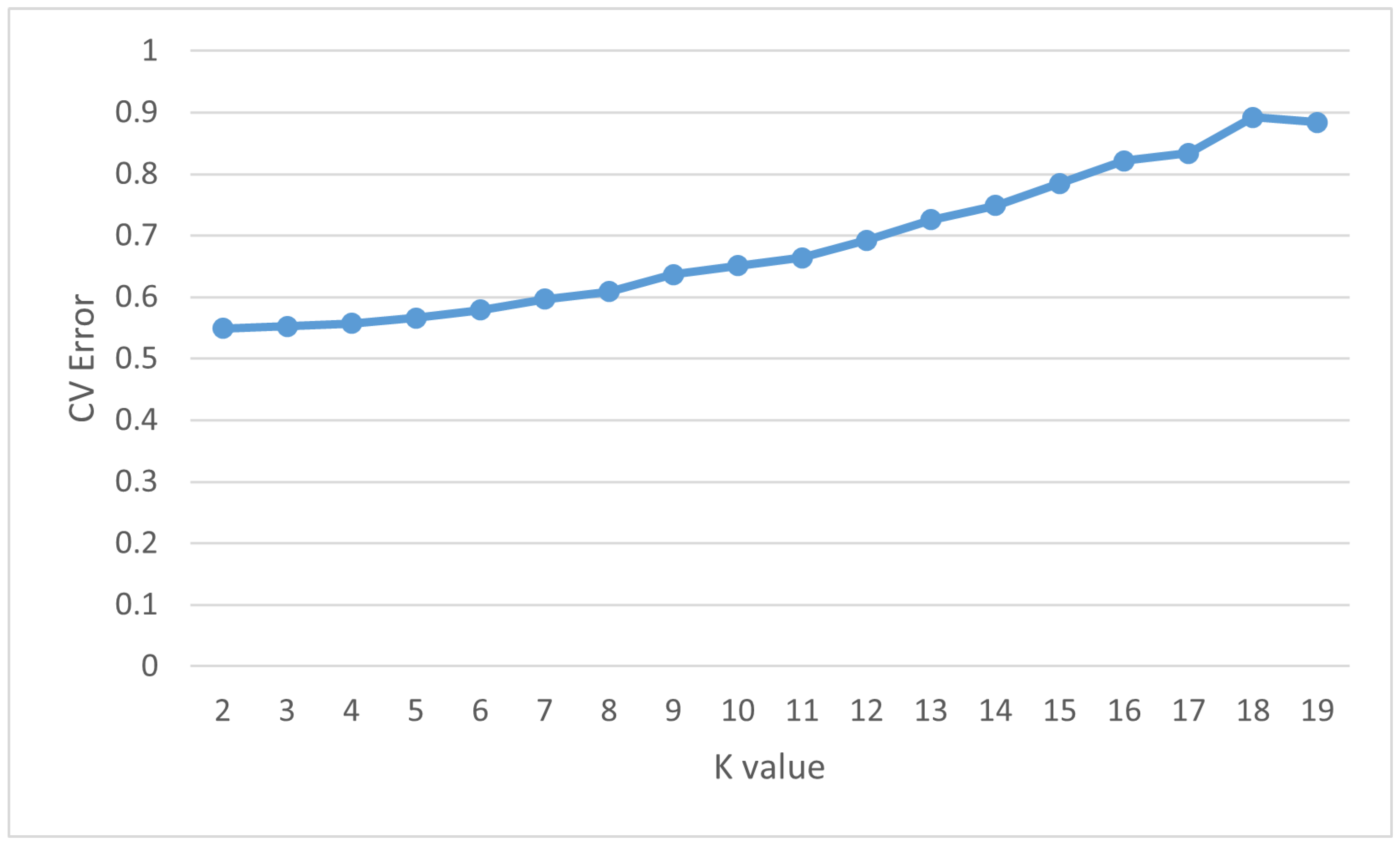
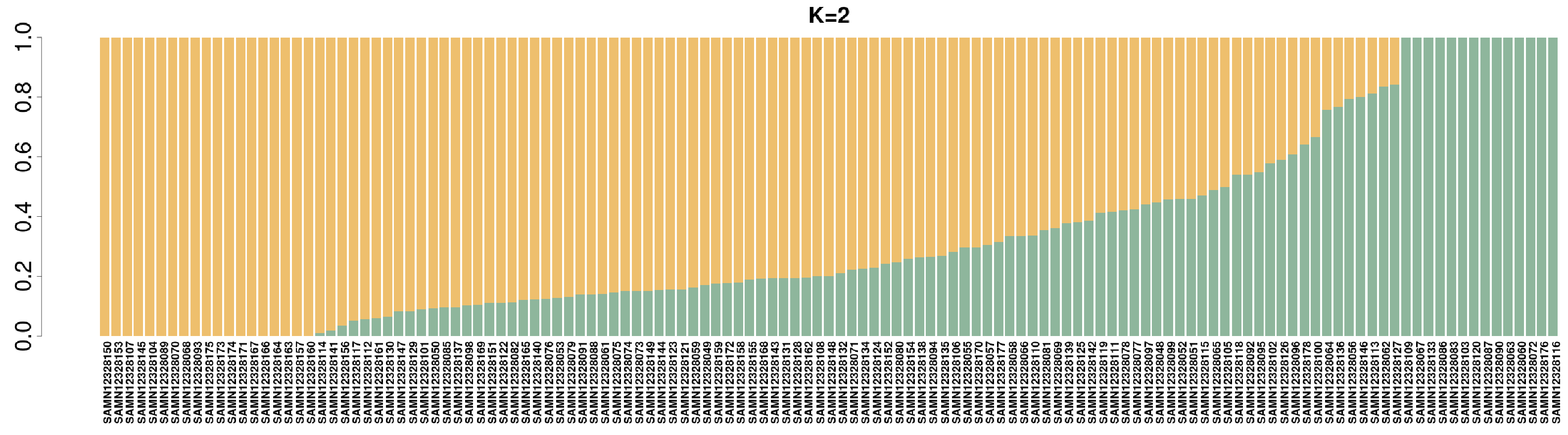
3.2. Population Structure
3.3. Inbreeding Analysis and Subgroup Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Qu, K.X.; Huang, B.Z.; Yang, G.R.; He, Z.X.; Zhang, Y.P.; Zan, L.S. Genetic diversity analysis of BMY cattle based on microsatellite DNA markers. Russ. J. Genet. 2012, 48, 435–441. [Google Scholar] [CrossRef]
- Xiaoting, X.; Kaixing, Q.; Fangyu, L.; Peng, J.; Qiuming, C.; Ningbo, C.; Jicai, Z.; Hong, C.; Bizhi, H.; Chuzhao, L. Abundant Genetic Diversity of Yunling Cattle Based on Mitochondrial Genome. Animals 2019, 9, 641. [Google Scholar] [CrossRef]
- Yan, B. Population Genomics Research on Chaidamu Yellow Cattle. Master’s Thesis, Lanzhou University, Lanzhou, China, 2019. [Google Scholar]
- Bravo, S.; Larama, G.; Quiñones, J.; Paz, E.; Rodero, E.; Sepúlveda, N. Genetic diversity and phylogenetic relationship among araucana creole sheep and Spanish sheep breeds. Small Rumin. Res. 2019, 172, 23–30. [Google Scholar] [CrossRef]
- Xianbo, J.; Peng, D.; Shiyi, C.; Shaokang, Z.; Jie, W.; Songjia, L. Analysis of MC1R, MITF, TYR, TYRP1, and MLPH Genes Polymorphism in Four Rabbit Breeds with Different Coat Colors. Animals 2021, 11, 81. [Google Scholar] [CrossRef]
- Qu, K.; Zhang, J.; Yang, G.; He, Z.; Jin, X.; Wang, A.; Yuan, X.; Huang, B.; Zan, L. Karyotypic analysis of BMY cattle and Brahman. J. Northwest A & F Univ.-Nat. Sci. Ed. 2011, 39, 8–12. [Google Scholar] [CrossRef]
- Xia, X.; Yao, Y.; Li, C.; Zhang, F.; Qu, K.; Chen, H.; Huang, B.; Lei, C. Genetic diversity of Chinese cattle revealed by Y-SNP and Y-STR markers. Anim. Genet. 2019, 50, 64–69. [Google Scholar] [CrossRef]
- Chen, Q.; Zhan, J.; Shen, J.; Qu, K.; Hanif, Q.; Liu, J.; Zhang, J.; Chen, N.; Chen, H.; Huang, B.; et al. Whole-genome resequencing reveals diversity, global and local ancestry proportions in yunling cattle. J. Anim. Breed. Genet. 2020, 137, 641–650. [Google Scholar] [CrossRef]
- Sambrook, J.; Russell, D.W.; Irwin, C.A.; Janssen, K.A. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2001; p. 1890. [Google Scholar]
- Deorowicz, S.; Kokot, M.; Grabowski, S.; Debudaj-Grabysz, A. KMC 2: Fast and resource-frugal k-mer counting. Bioinformatics 2015, 31, 1569–1576. [Google Scholar] [CrossRef]
- Vurture, G.W.; Sedlazeck, F.J.; Nattestad, M.; Underwood, C.J.; Fang, H.; Gurtowski, J.; Schatz, M.C. GenomeScope: Fast reference-free genome profiling from short reads. Bioinformatics 2017, 33, 2202–2204. [Google Scholar] [CrossRef]
- Servant, N.; Varoquaux, N.; Lajoie, B.R.; Viara, E.; Chen, C.J.; Vert, J.P.; Heard, E.; Dekker, J.; Barillot, E. HiC-Pro: An optimized and flexible pipeline for Hi-C data processing. Genome Biol. 2015, 16, 259. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2011, 9, 357–359. [Google Scholar] [CrossRef]
- Burton, J.N.; Adey, A.; Patwardhan, R.P.; Qiu, R.; Kitzman, J.O.; Shendure, J. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat. Biotechnol. 2013, 31, 1119–1125. [Google Scholar] [CrossRef] [PubMed]
- Kaymak, I.; Luda, K.M.; Duimstra, L.R.; Ma, E.H.; Longo, J.; Dahabieh, M.S.; Faubert, B.; Oswald, B.M.; Watson, M.J.; Kitchen-Goosen, S.M.; et al. Carbon source availability drives nutrient utilization in CD8+ T cells. Cell Metab. 2022, 34, 1298–1311.e6. [Google Scholar] [CrossRef]
- Zdobnov, E.M.; Apweiler, R. InterProScan—An integration platform for the signature-recognition methods in InterPro. Bioinformatics 2001, 17, 847–848. [Google Scholar] [CrossRef]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A Program for Improved Detection of Transfer RNA Genes in Genomic Sequence. Bioinformatics 1997, 25, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Griffiths-Jones, S.; Moxon, S.; Marshall, M.; Khanna, A.; Eddy, S.R.; Bateman, A. Rfam: Annotating non-coding RNAs in complete genomes. Bioinformatics 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Lagesen, K.; Hallin, P.; Rødland, E.A.; Stærfeldt, H.H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Bioinformatics 1997, 25, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Nekrutenko, A.; Taylor, J. Next-generation sequencing data interpretation: Enhancing reproducibility and accessibility. Nat. Rev. Genet. 2012, 13, 667–672. [Google Scholar] [CrossRef]
- Wei, Z. The Assembled Draft Genome of Yunling Cattle Annotation Results of Repeated Sequences, Gene Structure and Functional Prediction. Available online: https://figshare.com/articles/dataset/The_assembled_draft_genome_of_Yunling_cattle_annotation_results_of_repeated_sequences_gene_structure_and_functional_prediction_/23391614/1 (accessed on 20 October 2023).
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Zhang, C.; Dong, S.S.; Xu, J.Y.; He, W.M.; Yang, T.L. PopLDdecay: A fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Nucleic Acids Res. 2019, 35, 1786–1788. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.H.; Lange, K. Enhancements to the ADMIXTURE algorithm for individual ancestry estimation. BMC Bioinform. 2011, 12, 246. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Sun, H.; Lai, W.; Hu, M.; Zhang, Y.; Bai, C.; Liu, J.; Ren, H.; Li, F.; Yan, S. Genome-wide re-sequencing reveals population structure and genetic diversity of Bohai Black cattle. Anim. Genet. 2022, 53, 133–136. [Google Scholar] [CrossRef]
- Strucken, E.M.; Gebrehiwot, N.Z.; Swaminathan, M.; Joshi, S.; Kalaldeh, M.A.; Gibson, J.P. Genetic diversity and effective population sizes of thirteen Indian cattle breeds. Genet. Sel. Evol. 2021, 53, 559–575. [Google Scholar] [CrossRef]
- Edwards, C.E.; Tessier, B.C.; Swift, J.F.; Bassüner, B.; Linan, A.G.; Albrecht, M.A.; Yatskievych, G.A. Conservation genetics of the threatened plant species Physaria filiformis (Missouri bladderpod) reveals strong genetic structure and a possible cryptic species. PLoS ONE 2021, 16, e0247586. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Breed | Category | Area | Size | Pi | (()/) | |||
|---|---|---|---|---|---|---|---|---|
| Yunling | B. indicus | China | 129 | 0.000166 | 0.177 | 0.256 | 0.251 | 0.309 |
| Bohai Black | Hybrid | China | 40 | 0.000334 | 0.191 | 0.259 | 0.313 | 0.263 |
| Dabieshan | Hybrid | China | 4 | 0.000404 | 0.252 | 0.243 | 0.083 | −0.037 |
| Jiaxian Red | Hybrid | China | 5 | 0.000389 | 0.252 | 0.247 | 0.086 | −0.02 |
| Lingnan | Hybrid | China | 8 | 0.000392 | 0.235 | 0.255 | 0.145 | 0.078 |
| Luxi | Hybrid | China | 7 | 0.000371 | 0.282 | 0.236 | 0.026 | −0.195 |
| Nanyang | Hybrid | China | 7 | 0.000433 | 0.317 | 0.279 | 0.154 | −0.136 |
| Zaobei | Hybrid | China | 5 | 0.0004 | 0.262 | 0.247 | 0.046 | −0.06 |
| Guangfeng | Bos indicus | China | 4 | 0.000336 | 0.203 | 0.188 | 0.26 | −0.08 |
| Ji’an | B. indicus | China | 4 | 0.000325 | 0.195 | 0.176 | 0.29 | −0.108 |
| Jinjiang | B. indicus | China | 4 | 0.000368 | 0.241 | 0.217 | 0.123 | −0.111 |
| Leiqiong | B. indicus | China | 3 | 0.000321 | 0.176 | 0.153 | 0.358 | −0.15 |
| Wannan | B. indicus | China | 6 | 0.000302 | 0.193 | 0.174 | 0.298 | −0.109 |
| Kazakh | Bos taurus | China | 11 | 0.000356 | 0.232 | 0.236 | 0.154 | 0.017 |
| Mongolian | B. taurus | China | 9 | 0.000369 | 0.243 | 0.241 | 0.115 | −0.008 |
| Xuanhan | B. taurus | China | 5 | 0.000395 | 0.262 | 0.246 | 0.044 | −0.065 |
| Charolais | B. taurus | Europe | 10 | 0.00034 | 0.22 | 0.221 | 0.199 | 0.005 |
| Jersey | B. taurus | Europe | 20 | 0.000281 | 0.186 | 0.187 | 0.326 | 0.005 |
| Limousin | B. taurus | Europe | 10 | 0.000327 | 0.221 | 0.213 | 0.197 | −0.038 |
| Simmental | B. taurus | Europe | 30 | 0.000327 | 0.22 | 0.22 | 0.199 | 0 |
| Hanwoo | B. taurus | South Korea | 11 | 0.000328 | 0.22 | 0.214 | 0.199 | −0.028 |
| Mishima-Ushi | B. taurus | Japan | 8 | 0.0002 | 0.094 | 0.089 | 0.657 | −0.056 |
| Angus | B. taurus | Europe | 33 | 0.000309 | 0.208 | 0.208 | 0.242 | 0 |
| Hereford | B. taurus | Europe | 21 | 0.000288 | 0.191 | 0.194 | 0.309 | 0.015 |
| Holstein | B. taurus | Europe | 20 | 0.000301 | 0.2 | 0.201 | 0.273 | 0.005 |
| Brahman | B. indicus | India | 5 | 0.000268 | 0.143 | 0.173 | 0.47 | 0.173 |
| Group | (( − )/) | |||
|---|---|---|---|---|
| Subgroup I | 0.175 | 0.254 | 0.23 | 0.311 |
| Subgroup II | 0.177 | 0.256 | 0.246 | 0.309 |
| Yunling cattle | 0.177 | 0.256 | 0.251 | 0.309 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, J.; Zhang, L.; Gao, L.; Wei, Z.; Dang, D.; Yang, L. Population Structure and Genetic Diversity of Yunling Cattle Determined by Whole-Genome Resequencing. Genes 2023, 14, 2141. https://doi.org/10.3390/genes14122141
Chen J, Zhang L, Gao L, Wei Z, Dang D, Yang L. Population Structure and Genetic Diversity of Yunling Cattle Determined by Whole-Genome Resequencing. Genes. 2023; 14(12):2141. https://doi.org/10.3390/genes14122141
Chicago/Turabian StyleChen, Jian, Lilian Zhang, Lutao Gao, Zaichao Wei, Dong Dang, and Linnan Yang. 2023. "Population Structure and Genetic Diversity of Yunling Cattle Determined by Whole-Genome Resequencing" Genes 14, no. 12: 2141. https://doi.org/10.3390/genes14122141