
The Landscape of Monogenic Parkinson’s Disease in Populations of Non-European Ancestry: A Narrative Review
 ,
,  , ,
, ,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
3. Results
3.1. Autosomal-Dominant Genes
3.1.1. LRRK2
3.1.2. SNCA
3.1.3. VPS35
3.2. Autosomal-Recessive Genes
3.2.1. PRKN (Parkin)
3.2.2. PINK1
3.2.3. DJ-1
3.3. GBA1
3.4. Other Rare Genes
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Koros, C.; Simitsi, A.; Stefanis, L. Genetics of Parkinson’s Disease: Genotype-Phenotype Correlations. Int. Rev. Neurobiol. 2017, 132, 197–231. [Google Scholar] [CrossRef]
- Prendes Fernandez, P.; Blazquez Estrada, M.; Sol Alvarez, J.; Alvarez Martinez, V.; Suarez San Martin, E.; Garcia Fernandez, C.; Alvarez Carriles, J.C.; Lozano Aragoneses, B.; Saiz Ayala, A.; Santamarta Liebana, E.; et al. Analysis of deep brain stimulation of the subthalamic nucleus (STN-DBS) in patients with monogenic PRKN and LRRK2 forms of Parkinson’s disease. Park. Relat. Disord. 2023, 107, 105282. [Google Scholar] [CrossRef]
- Salles, P.A.; Liao, J.; Shuaib, U.; Mata, I.F.; Fernandez, H.H. A Review on Response to Device-Aided Therapies Used in Monogenic Parkinsonism and GBA Variants Carriers: A Need for Guidelines and Comparative Studies. J. Park. Dis. 2022, 12, 1703–1725. [Google Scholar] [CrossRef] [PubMed]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the α-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef]
- Schumacher-Schuh, A.F.; Bieger, A.; Okunoye, O.; Mok, K.Y.; Lim, S.Y.; Bardien, S.; Ahmad-Annuar, A.; Santos-Lobato, B.L.; Strelow, M.Z.; Salama, M.; et al. Underrepresented Populations in Parkinson’s Genetics Research: Current Landscape and Future Directions. Mov. Disord. 2022, 37, 1593–1604. [Google Scholar] [CrossRef] [PubMed]
- Paisan-Ruiz, C.; Jain, S.; Evans, E.W.; Gilks, W.P.; Simon, J.; van der Brug, M.; Lopez de Munain, A.; Aparicio, S.; Gil, A.M.; Khan, N.; et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron 2004, 44, 595–600. [Google Scholar] [CrossRef]
- Roosen, D.A.; Cookson, M.R. LRRK2 at the interface of autophagosomes, endosomes and lysosomes. Mol. Neurodegener. 2016, 11, 73. [Google Scholar] [CrossRef]
- Healy, D.G.; Falchi, M.; O’Sullivan, S.S.; Bonifati, V.; Durr, A.; Bressman, S.; Brice, A.; Aasly, J.; Zabetian, C.P.; Goldwurm, S.; et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: A case-control study. Lancet. Neurol. 2008, 7, 583–590. [Google Scholar] [CrossRef]
- Funayama, M.; Hasegawa, K.; Ohta, E.; Kawashima, N.; Komiyama, M.; Kowa, H.; Tsuji, S.; Obata, F. An LRRK2 mutation as a cause for the parkinsonism in the original PARK8 family. Ann. Neurol. 2005, 57, 918–921. [Google Scholar] [CrossRef]
- Alcalay, R.N.; Mejia-Santana, H.; Mirelman, A.; Saunders-Pullman, R.; Raymond, D.; Palmese, C.; Caccappolo, E.; Ozelius, L.; Orr-Urtreger, A.; Clark, L.; et al. Neuropsychological performance in LRRK2 G2019S carriers with Parkinson’s disease. Park. Relat. Disord. 2015, 21, 106–110. [Google Scholar] [CrossRef]
- Ehrminger, M.; Leu-Semenescu, S.; Cormier, F.; Corvol, J.C.; Vidailhet, M.; Debellemaniere, E.; Brice, A.; Arnulf, I. Sleep aspects on video-polysomnography in LRRK2 mutation carriers. Mov. Disord. 2015, 30, 1839–1843. [Google Scholar] [CrossRef] [PubMed]
- Lesage, S.; Durr, A.; Tazir, M.; Lohmann, E.; Leutenegger, A.L.; Janin, S.; Pollak, P.; Brice, A.; French Parkinson’s Disease Genetics Study, G. LRRK2 G2019S as a cause of Parkinson’s disease in North African Arabs. N. Engl. J. Med. 2006, 354, 422–423. [Google Scholar] [CrossRef]
- Ozelius, L.J.; Senthil, G.; Saunders-Pullman, R.; Ohmann, E.; Deligtisch, A.; Tagliati, M.; Hunt, A.L.; Klein, C.; Henick, B.; Hailpern, S.M.; et al. LRRK2 G2019S as a cause of Parkinson’s disease in Ashkenazi Jews. N. Engl. J. Med. 2006, 354, 424–425. [Google Scholar] [CrossRef] [PubMed]
- Bozi, M.; Papadimitriou, D.; Antonellou, R.; Moraitou, M.; Maniati, M.; Vassilatis, D.K.; Papageorgiou, S.G.; Leonardos, A.; Tagaris, G.; Malamis, G.; et al. Genetic assessment of familial and early-onset Parkinson’s disease in a Greek population. Eur. J. Neurol. 2014, 21, 963–968. [Google Scholar] [CrossRef] [PubMed]
- Kaiyrzhanov, R.; Rizig, M.; Aitkulova, A.; Zharkinbekova, N.; Shashkin, C.; Kaishibayeva, G.; Karimova, A.; Khaibullin, T.; Sadykova, D.; Ganieva, M.; et al. Parkinson’s Disease in Central Asian and Transcaucasian Countries: A Review of Epidemiology, Genetics, Clinical Characteristics, and Access to Care. Park. Dis. 2019, 2019, 2905739. [Google Scholar] [CrossRef]
- Kaiyrzhanov, R.; Aitkulova, A.; Vandrovcova, J.; Murphy, D.; Zharkinbekova, N.; Shashkin, C.; Akhmetzhanov, V.; Kaishibayeva, G.; Karimova, A.; Myrzayev, Z.; et al. A glimpse of the genetics of young-onset Parkinson’s disease in Central Asia. Mol. Genet. Genom. Med. 2021, 9, e1671. [Google Scholar] [CrossRef]
- Li, Y.; Ikeda, A.; Yoshino, H.; Oyama, G.; Kitani, M.; Daida, K.; Hayashida, A.; Ogaki, K.; Yoshida, K.; Kimura, T.; et al. Clinical characterization of patients with leucine-rich repeat kinase 2 genetic variants in Japan. J. Hum. Genet. 2020, 65, 771–781. [Google Scholar] [CrossRef]
- Shu, L.; Zhang, Y.; Sun, Q.; Pan, H.; Tang, B. A Comprehensive Analysis of Population Differences in LRRK2 Variant Distribution in Parkinson’s Disease. Front. Aging Neurosci. 2019, 11, 13. [Google Scholar] [CrossRef]
- Kanaya, Y.; Kume, K.; Morino, H.; Ohsawa, R.; Kurashige, T.; Kamada, M.; Torii, T.; Izumi, Y.; Maruyama, H.; Kawakami, H. Analysis of genetic risk factors in Japanese patients with Parkinson’s disease. J. Hum. Genet. 2021, 66, 957–964. [Google Scholar] [CrossRef]
- Do, M.D.; Tran, T.N.; Luong, A.B.; Le, L.H.G.; Van Le, T.; Le, K.T.; Van Vo, N.T.; Le, T.N.; Vu, H.A.; Mai, T.P. Clinical and genetic analysis of Vietnamese patients diagnosed with early-onset Parkinson’s disease. Brain Behav. 2023, 13, e2950. [Google Scholar] [CrossRef]
- Pulkes, T.; Papsing, C.; Thakkinstian, A.; Pongpakdee, S.; Kulkantrakorn, K.; Hanchaiphiboolkul, S.; Tiamkao, S.; Boonkongchuen, P. Confirmation of the association between LRRK2 R1628P variant and susceptibility to Parkinson’s disease in the Thai population. Park. Relat. Disord. 2014, 20, 1018–1021. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Qin, L.; Pan, H.; Liu, Z.; Jiang, L.; He, Y.; Zeng, Q.; Zhou, X.; Zhou, X.; Zhou, Y.; et al. The role of genetics in Parkinson’s disease: A large cohort study in Chinese mainland population. Brain A J. Neurol. 2020, 143, 2220–2234. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.M.; Zhou, X.Y.; Liang, X.N.; Lin, J.R.; Xu, Y.D.; Chen, C.; Wei, S.D.; Chen, Q.S.; Liu, F.T.; Zhao, J.; et al. The genetic spectrum of a cohort of patients clinically diagnosed as Parkinson’s disease in mainland China. NPJ Park. Dis. 2023, 9, 76. [Google Scholar] [CrossRef] [PubMed]
- Vishwanathan Padmaja, M.; Jayaraman, M.; Srinivasan, A.V.; Srikumari Srisailapathy, C.R.; Ramesh, A. The SNCA (A53T, A30P, E46K) and LRRK2 (G2019S) mutations are rare cause of Parkinson’s disease in South Indian patients. Park. Relat. Disord. 2012, 18, 801–802. [Google Scholar] [CrossRef]
- Sadhukhan, D.; Biswas, A.; Bhaduri, A.; Sarkar, N.; Biswas, A.; Das, S.K.; Banerjee, T.K.; Ray, K.; Ray, J. Role of LRRK2 variant p.Gly2019Ser in patients with Parkinsonism. Indian J. Med. Res. 2020, 151, 592–597. [Google Scholar] [CrossRef]
- Kishore, A.; Ashok Kumar Sreelatha, A.; Sturm, M.; von-Zweydorf, F.; Pihlstrom, L.; Raimondi, F.; Russell, R.; Lichtner, P.; Banerjee, M.; Krishnan, S.; et al. Understanding the role of genetic variability in LRRK2 in Indian population. Mov. Disord. 2019, 34, 496–505. [Google Scholar] [CrossRef]
- Okubadejo, N.U.; Rizig, M.; Ojo, O.O.; Jonvik, H.; Oshinaike, O.; Brown, E.; Houlden, H. Leucine rich repeat kinase 2 (LRRK2) GLY2019SER mutation is absent in a second cohort of Nigerian Africans with Parkinson disease. PLoS ONE 2018, 13, e0207984. [Google Scholar] [CrossRef]
- Rizig, M.; Ojo, O.O.; Athanasiou-Fragkouli, A.; Agabi, O.P.; Oshinaike, O.O.; Houlden, H.; Okubadejo, N.U. Negative screening for 12 rare LRRK2 pathogenic variants in a cohort of Nigerians with Parkinson’s disease. Neurobiol. Aging 2021, 99, 101.e15–101.e19. [Google Scholar] [CrossRef]
- Bardien, S.; Marsberg, A.; Keyser, R.; Lombard, D.; Lesage, S.; Brice, A.; Carr, J. LRRK2 G2019S mutation: Frequency and haplotype data in South African Parkinson’s disease patients. J. Neural Transm. 2010, 117, 847–853. [Google Scholar] [CrossRef]
- Cornejo-Olivas, M.; Torres, L.; Velit-Salazar, M.R.; Inca-Martinez, M.; Mazzetti, P.; Cosentino, C.; Micheli, F.; Perandones, C.; Dieguez, E.; Raggio, V.; et al. Variable frequency of LRRK2 variants in the Latin American research consortium on the genetics of Parkinson’s disease (LARGE-PD), a case of ancestry. NPJ Park. Dis. 2017, 3, 19. [Google Scholar] [CrossRef]
- Torrealba-Acosta, G.; Yu, E.; Lobo-Prada, T.; Ruiz-Martinez, J.; Gorostidi-Pagola, A.; Gan-Or, Z.; Carazo-Cespedes, K.; Trempe, J.F.; Mata, I.F.; Fornaguera-Trias, J. Clinical and Genetic Analysis of Costa Rican Patients with Parkinson’s Disease. Front. Neurol. 2021, 12, 656342. [Google Scholar] [CrossRef] [PubMed]
- Vekrellis, K.; Xilouri, M.; Emmanouilidou, E.; Rideout, H.J.; Stefanis, L. Pathological roles of α-synuclein in neurological disorders. Lancet Neurol. 2011, 10, 1015–1025. [Google Scholar] [CrossRef]
- Spira, P.J.; Sharpe, D.M.; Halliday, G.; Cavanagh, J.; Nicholson, G.A. Clinical and pathological features of a Parkinsonian syndrome in a family with an Ala53Thr α-synuclein mutation. Ann. Neurol. 2001, 49, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Zarranz, J.J.; Alegre, J.; Gomez-Esteban, J.C.; Lezcano, E.; Ros, R.; Ampuero, I.; Vidal, L.; Hoenicka, J.; Rodriguez, O.; Atares, B.; et al. The new mutation, E46K, of α-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 2004, 55, 164–173. [Google Scholar] [CrossRef]
- Kiely, A.P.; Asi, Y.T.; Kara, E.; Limousin, P.; Ling, H.; Lewis, P.; Proukakis, C.; Quinn, N.; Lees, A.J.; Hardy, J.; et al. α-Synucleinopathy associated with G51D SNCA mutation: A link between Parkinson’s disease and multiple system atrophy? Acta Neuropathol. 2013, 125, 753–769. [Google Scholar] [CrossRef]
- Papadimitriou, D.; Antonelou, R.; Miligkos, M.; Maniati, M.; Papagiannakis, N.; Bostantjopoulou, S.; Leonardos, A.; Koros, C.; Simitsi, A.; Papageorgiou, S.G.; et al. Motor and Nonmotor Features of Carriers of the p.A53T α-Synuclein Mutation: A Longitudinal Study. Mov. Disord. 2016, 31, 1226–1230. [Google Scholar] [CrossRef]
- Gwinn, K.; Devine, M.J.; Jin, L.W.; Johnson, J.; Bird, T.; Muenter, M.; Waters, C.; Adler, C.H.; Caselli, R.; Houlden, H.; et al. Clinical features, with video documentation, of the original familial lewy body parkinsonism caused by α-synuclein triplication (Iowa kindred). Mov. Disord.. 2011, 26, 2134–2136. [Google Scholar] [CrossRef] [PubMed]
- Nishioka, K.; Ross, O.A.; Ishii, K.; Kachergus, J.M.; Ishiwata, K.; Kitagawa, M.; Kono, S.; Obi, T.; Mizoguchi, K.; Inoue, Y.; et al. Expanding the clinical phenotype of SNCA duplication carriers. Mov. Disord. 2009, 24, 1811–1819. [Google Scholar] [CrossRef]
- Xiong, W.X.; Sun, Y.M.; Guan, R.Y.; Luo, S.S.; Chen, C.; An, Y.; Wang, J.; Wu, J.J. The heterozygous A53T mutation in the α-synuclein gene in a Chinese Han patient with Parkinson disease: Case report and literature review. J. Neurol. 2016, 263, 1984–1992. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.P.; Yu, S.H.; Zhang, G.H.; Hou, Y.B.; Gu, X.J.; Ou, R.W.; Shen, Y.; Song, W.; Chen, X.P.; Zhao, B.; et al. The mutation spectrum of Parkinson-disease-related genes in early-onset Parkinson’s disease in ethnic Chinese. Eur. J. Neurol. 2022, 29, 3218–3228. [Google Scholar] [CrossRef]
- Choi, J.M.; Woo, M.S.; Ma, H.I.; Kang, S.Y.; Sung, Y.H.; Yong, S.W.; Chung, S.J.; Kim, J.S.; Shin, H.W.; Lyoo, C.H.; et al. Analysis of PARK genes in a Korean cohort of early-onset Parkinson disease. Neurogenetics 2008, 9, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Itokawa, K.; Sekine, T.; Funayama, M.; Tomiyama, H.; Fukui, M.; Yamamoto, T.; Tamura, N.; Matsuda, H.; Hattori, N.; Araki, N. A case of α-synuclein gene duplication presenting with head-shaking movements. Mov. Disord. 2013, 28, 384–387. [Google Scholar] [CrossRef] [PubMed]
- Sekine, T.; Kagaya, H.; Funayama, M.; Li, Y.; Yoshino, H.; Tomiyama, H.; Hattori, N. Clinical course of the first Asian family with Parkinsonism related to SNCA triplication. Mov. Disord. 2010, 25, 2871–2875. [Google Scholar] [CrossRef]
- Kadakol, G.S.; Kulkarni, S.S.; Wali, G.M.; Gai, P.B. Molecular analysis of α-synuclein gene in Parkinson’s disease in North Karnataka, India. Neurol. India 2014, 62, 149–152. [Google Scholar] [CrossRef]
- Nagar, S.; Juyal, R.C.; Chaudhary, S.; Behari, M.; Gupta, M.; Rao, S.N.; Thelma, B.K. Mutations in the α-synuclein gene in Parkinson’s disease among Indians. Acta Neurol. Scand. 2001, 103, 120–122. [Google Scholar] [CrossRef] [PubMed]
- Yonova-Doing, E.; Atadzhanov, M.; Quadri, M.; Kelly, P.; Shawa, N.; Musonda, S.T.; Simons, E.J.; Breedveld, G.J.; Oostra, B.A.; Bonifati, V. Analysis of LRRK2, SNCA, Parkin, PINK1, and DJ-1 in Zambian patients with Parkinson’s disease. Park. Relat. Disord. 2012, 18, 567–571. [Google Scholar] [CrossRef]
- Mahne, A.C.; Carr, J.A.; Bardien, S.; Schutte, C.M. Clinical findings and genetic screening for copy number variation mutations in a cohort of South African patients with Parkinson’s disease. S. Afr. Med. J. Suid-Afr. Tydskr. Vir Geneeskd. 2016, 106, 623–625. [Google Scholar] [CrossRef]
- Milanowski, L.M.; Oshinaike, O.; Broadway, B.J.; Lindemann, J.A.; Soto-Beasley, A.I.; Walton, R.L.; Hanna Al-Shaikh, R.; Strongosky, A.J.; Fiesel, F.C.; Ross, O.A.; et al. Early-Onset Parkinson Disease Screening in Patients From Nigeria. Front. Neurol. 2020, 11, 594927. [Google Scholar] [CrossRef]
- Abreu, G.M.; Valenca, D.C.; Campos, M.J.; da Silva, C.P.; Pereira, J.S.; Araujo Leite, M.A.; Rosso, A.L.; Nicaretta, D.H.; Vasconcellos, L.F.; da Silva, D.J.; et al. Autosomal dominant Parkinson’s disease: Incidence of mutations in LRRK2, SNCA, VPS35 and GBA genes in Brazil. Neurosci. Lett. 2016, 635, 67–70. [Google Scholar] [CrossRef]
- Mohan, M.; Mellick, G.D. Role of the VPS35 D620N mutation in Parkinson’s disease. Park. Relat. Disord. 2017, 36, 10–18. [Google Scholar] [CrossRef]
- Struhal, W.; Presslauer, S.; Spielberger, S.; Zimprich, A.; Auff, E.; Bruecke, T.; Poewe, W.; Ransmayr, G.; Austrian, V.P.S.I.T. VPS35 Parkinson’s disease phenotype resembles the sporadic disease. J. Neural Transm. 2014, 121, 755–759. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.R.; Weissbach, A.; Heldmann, M.; Kasten, M.; Tunc, S.; Sue, C.M.; Svetel, M.; Kostic, V.S.; Segura-Aguilar, J.; Ramirez, A.; et al. Frequency of the D620N mutation in VPS35 in Parkinson disease. Arch. Neurol. 2012, 69, 1360–1364. [Google Scholar] [CrossRef]
- Ando, M.; Funayama, M.; Li, Y.; Kashihara, K.; Murakami, Y.; Ishizu, N.; Toyoda, C.; Noguchi, K.; Hashimoto, T.; Nakano, N.; et al. VPS35 mutation in Japanese patients with typical Parkinson’s disease. Mov. Disord. 2012, 27, 1413–1417. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.F.; Sun, Q.Y.; Lv, Z.Y.; Yu, R.L.; Li, K.; Zhang, Y.H.; Tian, J.Y.; Xia, K.; Yan, X.X.; Tang, B.S. VPS35 gene variants are not associated with Parkinson’s disease in the mainland Chinese population. Park. Relat. Disord. 2012, 18, 983–985. [Google Scholar] [CrossRef]
- Kumar, S.; Yadav, N.; Pandey, S.; Muthane, U.B.; Govindappa, S.T.; Abbas, M.M.; Behari, M.; Thelma, B.K. Novel and reported variants in Parkinson’s disease genes confer high disease burden among Indians. Park. Relat. Disord. 2020, 78, 46–52. [Google Scholar] [CrossRef]
- Sudhaman, S.; Behari, M.; Govindappa, S.T.; Muthane, U.B.; Juyal, R.C.; Thelma, B.K. VPS35 and EIF4G1 mutations are rare in Parkinson’s disease among Indians. Neurobiol. Aging 2013, 34, 2442.e1–2442.e3. [Google Scholar] [CrossRef]
- Blanckenberg, J.; Ntsapi, C.; Carr, J.A.; Bardien, S. EIF4G1 R1205H and VPS35 D620N mutations are rare in Parkinson’s disease from South Africa. Neurobiol. Aging 2014, 35, 445.e1–445.e3. [Google Scholar] [CrossRef] [PubMed]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef] [PubMed]
- van der Merwe, C.; Jalali Sefid Dashti, Z.; Christoffels, A.; Loos, B.; Bardien, S. Evidence for a common biological pathway linking three Parkinson’s disease-causing genes: Parkin, PINK1 and DJ-1. Eur. J. Neurosci. 2015, 41, 1113–1125. [Google Scholar] [CrossRef]
- Khan, N.L.; Graham, E.; Critchley, P.; Schrag, A.E.; Wood, N.W.; Lees, A.J.; Bhatia, K.P.; Quinn, N. Parkin disease: A phenotypic study of a large case series. Brain 2003, 126, 1279–1292. [Google Scholar] [CrossRef]
- Zhu, W.; Huang, X.; Yoon, E.; Bandres-Ciga, S.; Blauwendraat, C.; Billingsley, K.J.; Cade, J.H.; Wu, B.P.; Williams, V.H.; Schindler, A.B.; et al. Heterozygous PRKN mutations are common but do not increase the risk of Parkinson’s disease. Brain 2022, 145, 2077–2091. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.M.; Bounds, R.; Lincoln, S.; Hulihan, M.; Lin, C.H.; Hwu, W.L.; Chen, J.; Gwinn-Hardy, K.; Farrer, M. Parkin mutations and early-onset parkinsonism in a Taiwanese cohort. Arch. Neurol. 2005, 62, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.H.; Chen, P.L.; Tai, C.H.; Lin, H.I.; Chen, C.S.; Chen, M.L.; Wu, R.M. A clinical and genetic study of early-onset and familial parkinsonism in taiwan: An integrated approach combining gene dosage analysis and next-generation sequencing. Mov. Disord. 2019, 34, 506–515. [Google Scholar] [CrossRef] [PubMed]
- Tay, Y.W.; Tan, A.H.; Lim, J.L.; Lohmann, K.; Ibrahim, K.A.; Abdul Aziz, Z.; Chin, Y.T.; Mawardi, A.S.; Lim, T.T.; Looi, I.; et al. Genetic study of early-onset Parkinson’s disease in the Malaysian population. Park. Relat. Disord. 2023, 111, 105399. [Google Scholar] [CrossRef]
- Yoshino, H.; Li, Y.; Nishioka, K.; Daida, K.; Hayashida, A.; Ishiguro, Y.; Yamada, D.; Izawa, N.; Nishi, K.; Nishikawa, N.; et al. Genotype-phenotype correlation of Parkinson’s disease with PRKN variants. Neurobiol. Aging 2022, 114, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Pandey, S.; Tomar, L.R.; Kumar, S.; Dinesh, S.; Thelma, B.K. Expanding the canvas of PRKN mutations in familial and early-onset Parkinson disease. Park. Relat. Disord. 2019, 66, 216–219. [Google Scholar] [CrossRef]
- Chaudhary, S.; Behari, M.; Dihana, M.; Swaminath, P.V.; Govindappa, S.T.; Jayaram, S.; Goyal, V.; Maitra, A.; Muthane, U.B.; Juyal, R.C.; et al. Parkin mutations in familial and sporadic Parkinson’s disease among Indians. Park. Relat. Disord. 2006, 12, 239–245. [Google Scholar] [CrossRef]
- Biswas, A.; Gupta, A.; Naiya, T.; Das, G.; Neogi, R.; Datta, S.; Mukherjee, S.; Das, S.K.; Ray, K.; Ray, J. Molecular pathogenesis of Parkinson’s disease: Identification of mutations in the Parkin gene in Indian patients. Park. Relat. Disord. 2006, 12, 420–426. [Google Scholar] [CrossRef]
- Vinish, M.; Prabhakar, S.; Khullar, M.; Verma, I.; Anand, A. Genetic screening reveals high frequency of PARK2 mutations and reduced Parkin expression conferring risk for Parkinsonism in North West India. J. Neurol. Neurosurg. Psychiatry 2010, 81, 166–170. [Google Scholar] [CrossRef]
- Keyser, R.J.; Lombard, D.; Veikondis, R.; Carr, J.; Bardien, S. Analysis of exon dosage using MLPA in South African Parkinson’s disease patients. Neurogenetics 2010, 11, 305–312. [Google Scholar] [CrossRef]
- Haylett, W.L.; Keyser, R.J.; du Plessis, M.C.; van der Merwe, C.; Blanckenberg, J.; Lombard, D.; Carr, J.; Bardien, S. Mutations in the parkin gene are a minor cause of Parkinson’s disease in the South African population. Park. Relat. Disord. 2012, 18, 89–92. [Google Scholar] [CrossRef]
- Dekker, M.C.J.; Suleiman, J.M.; Bhwana, D.; Howlett, W.P.; Rashid, S.M.; van Minkelen, R.; Hamel, B.C. PRKN-related familial Parkinson’s disease: First molecular confirmation from East Africa. Park. Relat. Disord. 2020, 73, 14–15. [Google Scholar] [CrossRef] [PubMed]
- Monroy-Jaramillo, N.; Guerrero-Camacho, J.L.; Rodriguez-Violante, M.; Boll-Woehrlen, M.C.; Yescas-Gomez, P.; Alonso-Vilatela, M.E.; Lopez-Lopez, M. Genetic mutations in early-onset Parkinson’s disease Mexican patients: Molecular testing implications. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2014, 165B, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Guerrero Camacho, J.L.; Monroy Jaramillo, N.; Yescas Gomez, P.; Rodriguez Violante, M.; Boll Woehrlen, C.; Alonso Vilatela, M.E.; Lopez Lopez, M. High frequency of Parkin exon rearrangements in Mexican-mestizo patients with early-onset Parkinson’s disease. Mov. Disord. 2012, 27, 1047–1051. [Google Scholar] [CrossRef]
- Tipton, P.W.; Jaramillo-Koupermann, G.; Soto-Beasley, A.I.; Walton, R.L.; Soler-Rangel, S.; Romero-Osorio, O.; Diaz, C.; Moreno-Lopez, C.L.; Ross, O.A.; Wszolek, Z.K.; et al. Genetic characterization of Parkinson’s disease patients in Ecuador and Colombia. Park. Relat. Disord. 2020, 75, 27–29. [Google Scholar] [CrossRef]
- Marongiu, R.; Ferraris, A.; Ialongo, T.; Michiorri, S.; Soleti, F.; Ferrari, F.; Elia, A.E.; Ghezzi, D.; Albanese, A.; Altavista, M.C.; et al. PINK1 heterozygous rare variants: Prevalence, significance and phenotypic spectrum. Hum. Mutat. 2008, 29, 565. [Google Scholar] [CrossRef]
- Voigt, A.; Berlemann, L.A.; Winklhofer, K.F. The mitochondrial kinase PINK1: Functions beyond mitophagy. J. Neurochem. 2016, 139 (Suppl. 1), 232–239. [Google Scholar] [CrossRef] [PubMed]
- Ishihara-Paul, L.; Hulihan, M.M.; Kachergus, J.; Upmanyu, R.; Warren, L.; Amouri, R.; Elango, R.; Prinjha, R.K.; Soto, A.; Kefi, M.; et al. PINK1 mutations and parkinsonism. Neurology 2008, 71, 896–902. [Google Scholar] [CrossRef]
- Hayashida, A.; Li, Y.; Yoshino, H.; Daida, K.; Ikeda, A.; Ogaki, K.; Fuse, A.; Mori, A.; Takanashi, M.; Nakahara, T.; et al. The identified clinical features of Parkinson’s disease in homo-, heterozygous and digenic variants of PINK1. Neurobiol. Aging 2021, 97, 146.e1–146.e13. [Google Scholar] [CrossRef]
- Tan, E.K.; Yew, K.; Chua, E.; Puvan, K.; Shen, H.; Lee, E.; Puong, K.Y.; Zhao, Y.; Pavanni, R.; Wong, M.C.; et al. PINK1 mutations in sporadic early-onset Parkinson’s disease. Mov. Disord. 2006, 21, 789–793. [Google Scholar] [CrossRef]
- Youn, J.; Lee, C.; Oh, E.; Park, J.; Kim, J.S.; Kim, H.T.; Cho, J.W.; Park, W.Y.; Jang, W.; Ki, C.S. Genetic variants of PARK genes in Korean patients with early-onset Parkinson’s disease. Neurobiol. Aging 2019, 75, 224.e9–224.e15. [Google Scholar] [CrossRef] [PubMed]
- Caritativo, E.C.A.; Yu, J.R.T.; Bautista, J.M.P.; Nishioka, K.; Jamora, R.D.G.; Yalung, P.M.; Ng, A.R.; Hattori, N. Genetic screening of Filipinos suspected with familial Parkinson’s disease: A pilot study. Park. Relat. Disord. 2023, 108, 105319. [Google Scholar] [CrossRef]
- Biswas, A.; Sadhukhan, T.; Majumder, S.; Misra, A.K.; Das, S.K.; Variation Consortium, I.G.; Ray, K.; Ray, J. Evaluation of PINK1 variants in Indian Parkinson’s disease patients. Park. Relat. Disord. 2010, 16, 167–171. [Google Scholar] [CrossRef]
- Morales-Briceno, H.; Ong, T.L.; Duma, S.R.; Murray, N.; Pepper, E.M.; Ha, A.; Tchan, M.C.; Fung, V.S.C. Recurrent Biallelic p.L347P PINK1 Variant in Polynesians with Parkinsonism and Isolated Dopa-Responsive Dystonia. Mov. Disord. Clin. Pract. 2022, 9, 696–697. [Google Scholar] [CrossRef]
- Patel, S.G.; Buchanan, C.M.; Mulroy, E.; Simpson, M.; Reid, H.A.; Drake, K.M.; Merriman, M.E.; Phipps-Green, A.; Cadzow, M.; Merriman, T.R.; et al. Potential PINK1 Founder Effect in Polynesia Causing Early-Onset Parkinson’s Disease. Mov. Disord.. 2021, 36, 2199–2200. [Google Scholar] [CrossRef]
- Bonifati, V.; Rizzu, P.; van Baren, M.J.; Schaap, O.; Breedveld, G.J.; Krieger, E.; Dekker, M.C.; Squitieri, F.; Ibanez, P.; Joosse, M.; et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 2003, 299, 256–259. [Google Scholar] [CrossRef]
- Macedo, M.G.; Verbaan, D.; Fang, Y.; van Rooden, S.M.; Visser, M.; Anar, B.; Uras, A.; Groen, J.L.; Rizzu, P.; van Hilten, J.J.; et al. Genotypic and phenotypic characteristics of Dutch patients with early onset Parkinson’s disease. Mov. Disord.. 2009, 24, 196–203. [Google Scholar] [CrossRef]
- Guo, J.F.; Zhang, X.W.; Nie, L.L.; Zhang, H.N.; Liao, B.; Li, J.; Wang, L.; Yan, X.X.; Tang, B.S. Mutation analysis of Parkin, PINK1 and DJ-1 genes in Chinese patients with sporadic early onset parkinsonism. J. Neurol. 2010, 257, 1170–1175. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Mata, I.F.; Lin, C.H.; Tzen, K.Y.; Lincoln, S.J.; Bounds, R.; Lockhart, P.J.; Hulihan, M.M.; Farrer, M.J.; Wu, R.M. Genotype-phenotype correlates in Taiwanese patients with early-onset recessive Parkinsonism. Mov. Disord. 2009, 24, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Abbas, M.M.; Govindappa, S.T.; Sudhaman, S.; Thelma, B.K.; Juyal, R.C.; Behari, M.; Muthane, U.B. Early Onset Parkinson’s disease due to DJ1 mutations: An Indian study. Park. Relat. Disord. 2016, 32, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Sadhukhan, T.; Biswas, A.; Das, S.K.; Ray, K.; Ray, J. DJ-1 variants in Indian Parkinson’s disease patients. Dis. Markers 2012, 33, 127–135. [Google Scholar] [CrossRef]
- Sidransky, E.; Samaddar, T.; Tayebi, N. Mutations in GBA are associated with familial Parkinson disease susceptibility and age at onset. Neurology 2009, 73, 1424–1425. [Google Scholar] [CrossRef] [PubMed]
- Gan-Or, Z.; Giladi, N.; Rozovski, U.; Shifrin, C.; Rosner, S.; Gurevich, T.; Bar-Shira, A.; Orr-Urtreger, A. Genotype-phenotype correlations between GBA mutations and Parkinson disease risk and onset. Neurology 2008, 70, 2277–2283. [Google Scholar] [CrossRef]
- Brockmann, K.; Srulijes, K.; Pflederer, S.; Hauser, A.K.; Schulte, C.; Maetzler, W.; Gasser, T.; Berg, D. GBA-associated Parkinson’s disease: Reduced survival and more rapid progression in a prospective longitudinal study. Mov. Disord. 2015, 30, 407–411. [Google Scholar] [CrossRef] [PubMed]
- Brockmann, K.; Srulijes, K.; Hauser, A.K.; Schulte, C.; Csoti, I.; Gasser, T.; Berg, D. GBA-associated PD presents with nonmotor characteristics. Neurology 2011, 77, 276–280. [Google Scholar] [CrossRef]
- Yu, Z.; Wang, T.; Xu, J.; Wang, W.; Wang, G.; Chen, C.; Zheng, L.; Pan, L.; Gong, D.; Li, X.; et al. Mutations in the glucocerebrosidase gene are responsible for Chinese patients with Parkinson’s disease. J. Hum. Genet. 2015, 60, 85–90. [Google Scholar] [CrossRef]
- Ren, J.; Zhang, R.; Pan, C.; Xu, J.; Sun, H.; Hua, P.; Zhang, L.; Zhang, W.; Xu, P.; Ma, C.; et al. Prevalence and genotype-phenotype correlations of GBA-related Parkinson disease in a large Chinese cohort. Eur. J. Neurol. 2022, 29, 1017–1024. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.L.; Wu-Chou, Y.H.; Lai, S.C.; Chang, H.C.; Yeh, T.H.; Weng, Y.H.; Chen, R.S.; Huang, Y.Z.; Lu, C.S. Contribution of glucocerebrosidase mutation in a large cohort of sporadic Parkinson’s disease in Taiwan. Eur. J. Neurol. 2011, 18, 1227–1232. [Google Scholar] [CrossRef]
- Mitsui, J.; Mizuta, I.; Toyoda, A.; Ashida, R.; Takahashi, Y.; Goto, J.; Fukuda, Y.; Date, H.; Iwata, A.; Yamamoto, M.; et al. Mutations for Gaucher disease confer high susceptibility to Parkinson disease. Arch. Neurol. 2009, 66, 571–576. [Google Scholar] [CrossRef]
- Lim, J.L.; Lohmann, K.; Tan, A.H.; Tay, Y.W.; Ibrahim, K.A.; Abdul Aziz, Z.; Mawardi, A.S.; Puvanarajah, S.D.; Lim, T.T.; Looi, I.; et al. Glucocerebrosidase (GBA) gene variants in a multi-ethnic Asian cohort with Parkinson’s disease: Mutational spectrum and clinical features. J. Neural Transm. 2022, 129, 37–48. [Google Scholar] [CrossRef]
- Pulkes, T.; Choubtum, L.; Chitphuk, S.; Thakkinstian, A.; Pongpakdee, S.; Kulkantrakorn, K.; Hanchaiphiboolkul, S.; Tiamkao, S.; Boonkongchuen, P. Glucocerebrosidase mutations in Thai patients with Parkinson’s disease. Park. Relat. Disord. 2014, 20, 986–991. [Google Scholar] [CrossRef]
- Kukkle, P.L.; Geetha, T.S.; Chaudhary, R.; Sathirapongsasuti, J.F.; Goyal, V.; Kandadai, R.M.; Kumar, H.; Borgohain, R.; Mukherjee, A.; Oliver, M.; et al. Genome-Wide Polygenic Score Predicts Large Number of High Risk Individuals in Monogenic Undiagnosed Young Onset Parkinson’s Disease Patients from India. Adv. Biol. 2022, 6, e2101326. [Google Scholar] [CrossRef] [PubMed]
- Biswas, A.; Sadhukhan, D.; Biswas, A.; Das, S.K.; Banerjee, T.K.; Bal, P.S.; Pal, S.; Ghosh, A.; Ray, K.; Ray, J. Identification of GBA mutations among neurodegenerative disease patients from eastern India. Neurosci. Lett. 2021, 751, 135816. [Google Scholar] [CrossRef] [PubMed]
- Mahungu, A.C.; Anderson, D.G.; Rossouw, A.C.; van Coller, R.; Carr, J.A.; Ross, O.A.; Bardien, S. Screening of the glucocerebrosidase (GBA) gene in South Africans of African ancestry with Parkinson’s disease. Neurobiol. Aging 2020, 88, 156.e11–156.e14. [Google Scholar] [CrossRef] [PubMed]
- Milanowski, L.M.; Oshinaike, O.; Walton, R.L.; Soto-Beasley, A.I.; Hanna Al-Shaikh, R.; Strongosky, A.J.; Ross, O.A.; Wszolek, Z.K.; Ogun, S.A. Screening of GBA Mutations in Nigerian Patients with Parkinson’s Disease. Mov. Disord. 2021, 36, 2971–2973. [Google Scholar] [CrossRef]
- Rizig, M.; Bandres-Ciga, S.; Makarious, M.B.; Ojo, O.O.; Crea, P.W.; Abiodun, O.V.; Levine, K.S.; Abubakar, S.A.; Achoru, C.O.; Vitale, D.; et al. Identification of genetic risk loci and causal insights associated with Parkinson’s disease in African and African admixed populations: A genome-wide association study. Lancet. Neurol. 2023, 22, 1015–1025. [Google Scholar] [CrossRef]
- Velez-Pardo, C.; Lorenzo-Betancor, O.; Jimenez-Del-Rio, M.; Moreno, S.; Lopera, F.; Cornejo-Olivas, M.; Torres, L.; Inca-Martinez, M.; Mazzetti, P.; Cosentino, C.; et al. The distribution and risk effect of GBA variants in a large cohort of PD patients from Colombia and Peru. Park. Relat. Disord. 2019, 63, 204–208. [Google Scholar] [CrossRef]
- Dos Santos, A.V.; Pestana, C.P.; Diniz, K.R.; Campos, M.; Abdalla-Carvalho, C.B.; de Rosso, A.L.; Pereira, J.S.; Nicaretta, D.H.; de Carvalho, W.L.; Dos Santos, J.M.; et al. Mutational analysis of GIGYF2, ATP13A2 and GBA genes in Brazilian patients with early-onset Parkinson’s disease. Neurosci. Lett. 2010, 485, 121–124. [Google Scholar] [CrossRef]
- Ravat, P.; Shinde, S.; Shinde, S.R.; Bangar, S.; Nayak, N.; Agarwal, P.A. Juvenile PLA2G6-Parkinsonism Due to Indian ‘Asian’ p.R741Q Mutation, and Response to STN DBS. Mov. Disord. 2022, 37, 657–658. [Google Scholar] [CrossRef]
- Li, N.N.; Wang, L.; Tan, E.K.; Cheng, L.; Sun, X.Y.; Lu, Z.J.; Li, J.Y.; Zhang, J.H.; Peng, R. Genetic analysis of CHCHD2 gene in Chinese Parkinson’s disease. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2016, 171, 1148–1152. [Google Scholar] [CrossRef]
- Global Parkinson’s Genetics Program. GP2: The Global Parkinson’s Genetics Program. Mov. Disord. 2021, 36, 842–851. [Google Scholar] [CrossRef] [PubMed]
- Lange, L.M.; Avenali, M.; Ellis, M.; Illarionova, A.; Keller Sarmiento, I.J.; Tan, A.H.; Madoev, H.; Galandra, C.; Junker, J.; Roopnarain, K.; et al. Elucidating causative gene variants in hereditary Parkinson’s disease in the Global Parkinson’s Genetics Program (GP2). NPJ Park. Dis. 2023, 9, 100. [Google Scholar] [CrossRef] [PubMed]
- Vollstedt, E.J.; Schaake, S.; Lohmann, K.; Padmanabhan, S.; Brice, A.; Lesage, S.; Tesson, C.; Vidailhet, M.; Wurster, I.; Hentati, F.; et al. Embracing Monogenic Parkinson’s Disease: The MJFF Global Genetic PD Cohort. Mov. Disord. 2023, 38, 286–303. [Google Scholar] [CrossRef] [PubMed]
- Zabetian, C.P.; Mata, I.F.; Latin American Research Consortium on the Genetics of PD. LARGE-PD: Examining the genetics of Parkinson’s disease in Latin America. Mov. Disord. 2017, 32, 1330–1331. [Google Scholar] [CrossRef]
- Sarihan, E.I.; Perez-Palma, E.; Niestroj, L.M.; Loesch, D.; Inca-Martinez, M.; Horimoto, A.; Cornejo-Olivas, M.; Torres, L.; Mazzetti, P.; Cosentino, C.; et al. Genome-Wide Analysis of Copy Number Variation in Latin American Parkinson’s Disease Patients. Mov. Disord. 2021, 36, 434–441. [Google Scholar] [CrossRef]
- Mok, K.Y.; East Asian Parkinson Disease Genomics, C. The East Asian Parkinson Disease Genomics Consortium. Lancet Neurol. 2021, 20, 982. [Google Scholar] [CrossRef]
- Rajan, R.; Divya, K.P.; Kandadai, R.M.; Yadav, R.; Satagopam, V.P.; Madhusoodanan, U.K.; Agarwal, P.; Kumar, N.; Ferreira, T.; Kumar, H.; et al. Genetic Architecture of Parkinson’s Disease in the Indian Population: Harnessing Genetic Diversity to Address Critical Gaps in Parkinson’s Disease Research. Front. Neurol. 2020, 11, 524. [Google Scholar] [CrossRef]
- Rizig, M.; Okubadejo, N.; Salama, M.; Thomas, O.; Akpalu, A.; Gouider, R.; Africa, I. The International Parkinson Disease Genomics Consortium Africa. Lancet Neurol. 2021, 20, 335. [Google Scholar] [CrossRef]


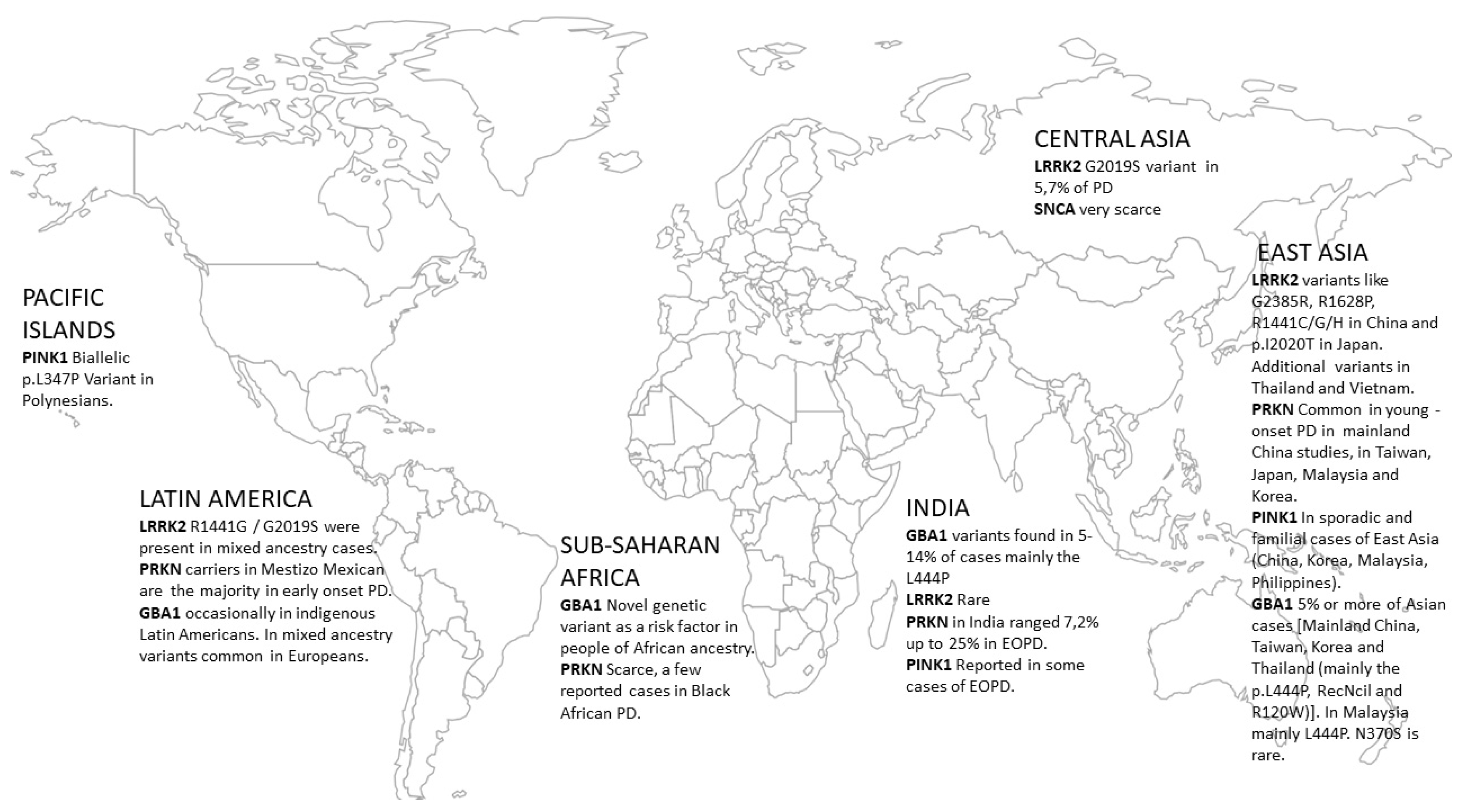{kind=link}
| Gene | East Asia (incl. China) | Central Asia | India | Sub-Saharan Africa | Latin America | Pacific Islands |
|---|---|---|---|---|---|---|
| α-synuclein (SNCA) | Very scarce; the p.A53T mutation reported on a different genetic background in Korea | Very scarce | N/A | N/A | N/A | N/A |
| Leucine Rich Repeat Kinase 2 (LRRK2) | Mainly region-specific variants like G2385R, R1628P. R1441C/G/H in China and p.I2020T in Japan. Additional variants in Thailand and Vietnam | G2019S in 5.7% of PD | Rare | Almost absent, only in mixed ancestry cases (South Africa) | R1441G/G2019S were present in mixed ancestry cases | |
| Vacuolar protein sorting 35 ortholog protein (VPS35) | The prevalence of VPS35 is higher among Japanese as compared to other Asians | N/A | Very Rare | N/A | N/A | N/A |
| Glucocerebrosidase gene variants (GBA1) | In total, 5% or more of Asian cases [Mainland China, Taiwan, Korea and Thailand (mainly the p.L444P, RecNcil and R120W)]. In Malaysia, mainly L444P. N370S is rare. | GBA1 variants found in 5–14% of cases, mainly the L444P and other variants (some rare in Europeans) | Novel genetic variant in GBA1 as a risk factor in people of African ancestry (present in 39% of cases). | Occasionally reported in indigenous Latin Americans, those of mixed ancestry had variants common in Europeans |
| Gene | East Asia (Incl. China) | Central Asia | India | Sub-Saharan Africa | Latin America | Pacific Islands |
|---|---|---|---|---|---|---|
| Parkin (PRKN) | Common in young-onset PD in mainland Chinese studies in Taiwan, Japan, Malaysia and Korea | Found in Kazakhstan PD patients | In India, PRKN mutations ranged from 7,2% up to 25% in EOPD | Scarce, a few reported cases in Black African PD | In a Mestizo Mexican population PRKN carriers are the majority in early-onset PD | |
| PTEN-induced kinase 1 (PINK1) | In sporadic and familial of East Asia (China, Korea, Malaysia, and Philippines) | Reported in some cases of early-onset PD | Rare | Biallelic p.L347P PINK1 Variant in Polynesians | ||
| Parkinson disease protein 7 (DJ-1) | Rather rare in East Asia (Malaysia, Mainland China, Taiwan, and Korea) | Rare cases | N/A |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koros, C.; Bougea, A.; Simitsi, A.M.; Papagiannakis, N.; Angelopoulou, E.; Pachi, I.; Antonelou, R.; Bozi, M.; Stamelou, M.; Stefanis, L. The Landscape of Monogenic Parkinson’s Disease in Populations of Non-European Ancestry: A Narrative Review. Genes 2023, 14, 2097. https://doi.org/10.3390/genes14112097
Koros C, Bougea A, Simitsi AM, Papagiannakis N, Angelopoulou E, Pachi I, Antonelou R, Bozi M, Stamelou M, Stefanis L. The Landscape of Monogenic Parkinson’s Disease in Populations of Non-European Ancestry: A Narrative Review. Genes. 2023; 14(11):2097. https://doi.org/10.3390/genes14112097
Chicago/Turabian StyleKoros, Christos, Anastasia Bougea, Athina Maria Simitsi, Nikolaos Papagiannakis, Efthalia Angelopoulou, Ioanna Pachi, Roubina Antonelou, Maria Bozi, Maria Stamelou, and Leonidas Stefanis. 2023. "The Landscape of Monogenic Parkinson’s Disease in Populations of Non-European Ancestry: A Narrative Review" Genes 14, no. 11: 2097. https://doi.org/10.3390/genes14112097