
DOCK3-Associated Neurodevelopmental Disorder—Clinical Features and Molecular Basis

Abstract
:1. Introduction
2. History of DOCK3-Deficiency
3. Clinical Features of DOCK3-Deficiency
4. Etiology and Molecular Pathways
4.1. DOCK3 Is Essential for Normal Cell Growth, Proliferation, and Migration
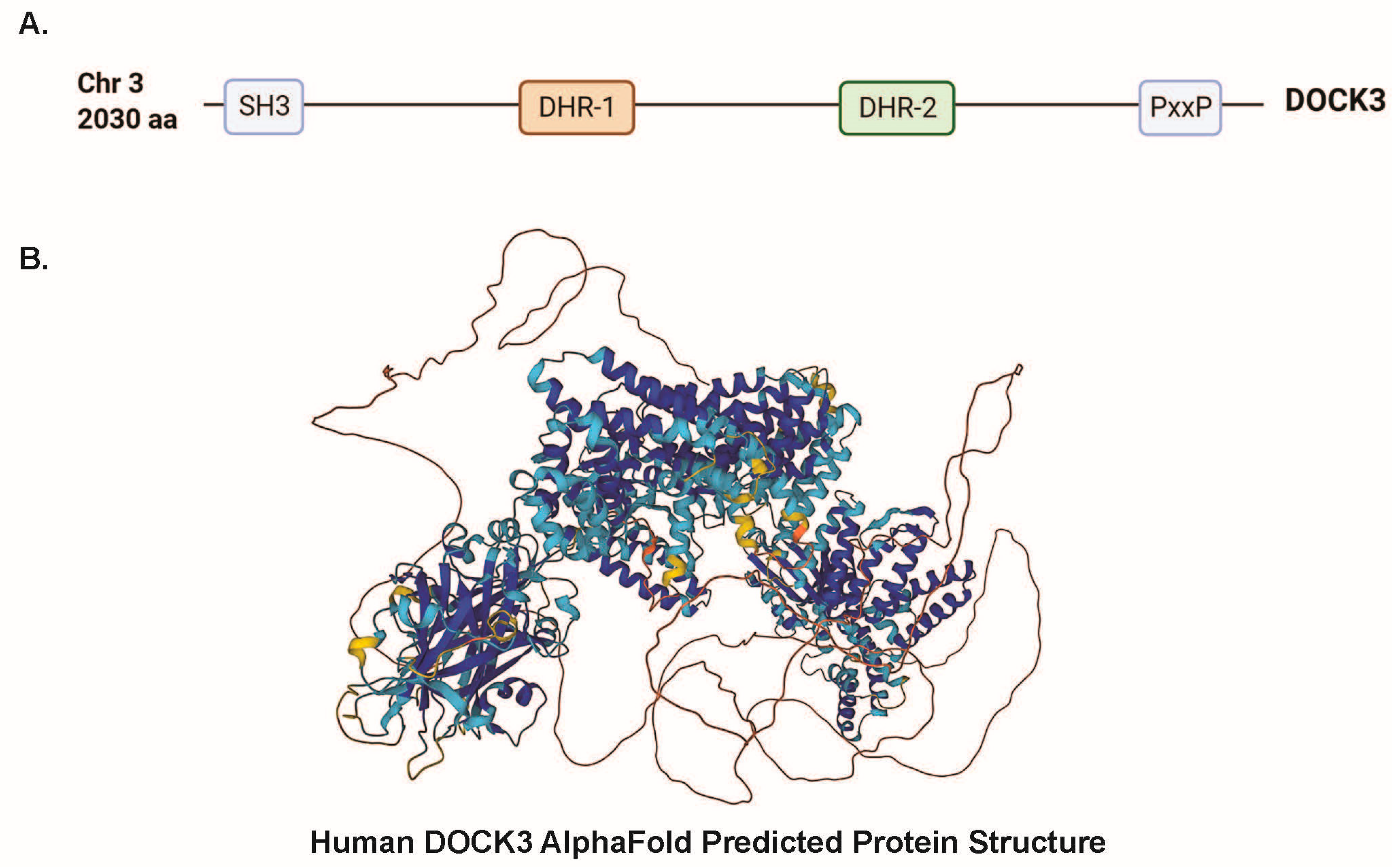
4.2. DOCK3 Is Comprises Key Evolutionary Conserved Domains Essential for Protein–Protein Interactions
4.3. Dock3-Deficient Mice Have Significant Developmental and Regenerative Defects
4.4. RAC1-Affected Pathways Affected by DOCK3 Disruption
4.5. Disease Phenotypes Associated with Deficient Functioning of Genes Interacting with DOCK3
5. Future Research
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Thapar, A.; Cooper, M.; Rutter, M. Neurodevelopmental disorders. Lancet Psychiatry 2017, 4, 339–346. [Google Scholar] [CrossRef]
- Velinov, M. Genomic Copy Number Variations in the Autism Clinic-Work in Progress. Front. Cell Neurosci. 2019, 13, 57. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Corominas, R.; Lin, G.N. De novo Mutations from Whole Exome Sequencing in Neurodevelopmental and Psychiatric Disorders: From Discovery to Application. Front. Genet. 2019, 10, 258. [Google Scholar] [CrossRef]
- Malpass, K. Neurodevelopmental disorders: Unlocking the secrets of autism through whole-exome sequencing. Nat. Rev. Neurol. 2012, 8, 295. [Google Scholar] [CrossRef]
- Wang, T.; Kim, C.N.; Bakken, T.E.; Gillentine, M.A.; Henning, B.; Mao, Y.; Gilissen, C.; SPARK Consortium; Nowakowski, T.J.; Eichler, E.E. Integrated gene analyses of de novo variants from 46,612 trios with autism and developmental disorders. Proc. Natl. Acad. Sci. USA 2022, 119, e2203491119. [Google Scholar] [CrossRef] [PubMed]
- Aslesh, T.; Yokota, T. Restoring SMN Expression: An Overview of the Therapeutic Developments for the Treatment of Spinal Muscular Atrophy. Cells 2022, 11, 417. [Google Scholar] [CrossRef]
- Canaud, G.; Gutierrez, J.C.L.; Irvine, A.D.; Vabres, P.; Hansford, J.R.; Ankrah, N.; Branle, F.; Papadimitriou, A.; Ridolfi, A.; O’Connell, P.; et al. Alpelisib for Treatment of Patients with PIK3CA-Related Overgrowth Spectrum (PROS). Genet. Med. 2023, 25, 100969. [Google Scholar] [CrossRef] [PubMed]
- Efron, D.; Delatycki, M.B.; de Silva, M.G.; Langbein, A.; Slaghuis, W.; Larson, A.; Dahl, H.H.; Forrest, S.M. A novel pericentric inversion of chromosome 3 cosegregates with a developmental-behavioral phenotype. J. Med. Genet. 2003, 40, E15. [Google Scholar] [CrossRef]
- de Silva, M.G.; Elliott, K.; Dahl, H.H.; Fitzpatrick, E.; Wilcox, S.; Delatycki, M.; Williamson, R.; Efron, D.; Lynch, M.; Forrest, S. Disruption of a novel member of a sodium/hydrogen exchanger family and DOCK3 is associated with an attention deficit hyperactivity disorder-like phenotype. J. Med. Genet. 2003, 40, 733–740. [Google Scholar] [CrossRef]
- Helbig, K.L.; Mroske, C.; Moorthy, D.; Sajan, S.A.; Velinov, M. Biallelic loss-of-function variants in DOCK3 cause muscle hypotonia, ataxia, and intellectual disability. Clin. Genet. 2017, 92, 430–433. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Peto, C.A.; Shelton, G.D.; Mizisin, A.; Sawchenko, P.E.; Schubert, D. Loss of Modifier of Cell Adhesion Reveals a Pathway Leading to Axonal Degeneration. J. Neurosci. 2009, 29, 118–130. [Google Scholar] [CrossRef]
- Iwata-Otsubo, A.; Ritter, A.L.; Weckselbatt, B.; Ryan, N.R.; Burgess, D.; Conlin, L.K.; Izumi, K. DOCK3-related neurodevelopmental syndrome: Biallelic intragenic deletion of DOCK3 in a boy with developmental delay and hypotonia. Am. J. Med. Genet. Part A 2018, 176, 241–245. [Google Scholar] [CrossRef]
- Wiltrout, K.; Ferrer, A.; van de Laar, I.; Namekata, K.; Harada, T.; Klee, E.W.; Zimmerman, M.T.; Cousin, M.A.; Kempainen, J.L.; Babovic-Vuksanovic, D.; et al. Variants in DOCK3 cause developmental delay and hypotonia. Eur. J. Hum. Genet. 2019, 27, 1225–1234. [Google Scholar] [CrossRef]
- Namekata, K.; Enokido, Y.; Iwasawa, K.; Kimura, H. MOCA Induces Membrane Spreading by Activating Rac1. J. Biol. Chem. 2004, 279, 14331–14337. [Google Scholar] [CrossRef]
- Chen, Q.; Chen, T.J.; Letourneau, P.C.; Costa Lda, F.; Schubert, D. Modifier of cell adhesion regulates N-cadherin-mediated cell-cell adhesion and neurite outgrowth. J. Neurosci. 2005, 25, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Namekata, K.; Harada, C.; Taya, C.; Guo, X.; Kimura, H.; Parada, L.F.; Harada, T. Dock3 induces axonal outgrowth by stimulating membrane recruitment of the WAVE complex. Proc. Natl. Acad. Sci. USA 2010, 107, 7586–7591. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Moreno, V.; Gadea, G.; Ahn, J.; Paterson, H.; Marra, P.; Pinner, S.; Sahai, E.; Marshall, C.J. Rac Activation and Inactivation Control Plasticity of Tumor Cell Movement. Cell 2008, 135, 510–523. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Mi, X.; Chen, L.; Jiang, G.; Wang, N.; Zhang, Y.; Deng, W.; Wang, Z.; Chen, G.; Wang, X. Dock3 Participate in Epileptogenesis through rac1 Pathway in Animal Models. Mol. Neurobiol. 2016, 53, 2715–2725. [Google Scholar] [CrossRef] [PubMed]
- Alexander, M.S.; Casar, J.C.; Motohashi, N.; Vieira, N.M.; Eisenberg, I.; Marshall, J.L.; Gasperini, M.J.; Lek, A.; Myers, J.A.; Estrella, E.A.; et al. MicroRNA-486–dependent modulation of DOCK3/PTEN/AKT signaling pathways improves muscular dystrophy–associated symptoms. J. Clin. Investig. 2014, 124, 2651–2667. [Google Scholar] [CrossRef]
- Reid, A.L.; Wang, Y.; Samani, A.; Hightower, R.M.; Lopez, M.A.; Gilbert, S.R.; Ianov, L.; Crossman, D.K.; Dell’Italia, L.J.; Millay, D.P.; et al. DOCK3 is a dosage-sensitive regulator of skeletal muscle and Duchenne muscular dystrophy-associated pathologies. Hum. Mol. Genet. 2020, 29, 2855–2871. [Google Scholar] [CrossRef] [PubMed]
- Makihara, S.; Morin, S.; Ferent, J.; Côté, J.-F.; Yam, P.T.; Charron, F. Polarized Dock Activity Drives Shh-Mediated Axon Guidance. Dev. Cell 2018, 46, 410–425.e417. [Google Scholar] [CrossRef] [PubMed]
- Namekata, K.; Watanabe, H.; Guo, X.; Kittaka, D.; Kawamura, K.; Kimura, A.; Harada, C.; Harada, T. Dock3 regulates BDNF-TrkB signaling for neurite outgrowth by forming a ternary complex with Elmo and RhoG. Genes Cells 2012, 17, 688–697. [Google Scholar] [CrossRef] [PubMed]
- Bai, N.; Hayashi, H.; Aida, T.; Namekata, K.; Harada, T.; Mishina, M.; Tanaka, K. Dock3 interaction with a glutamate-receptor NR2D subunit protects neurons from excitotoxicity. Mol. Brain 2013, 6, 22. [Google Scholar] [CrossRef]
- Samani, A.; Karuppasamy, M.; English, K.G.; Siler, C.A.; Wang, Y.; Widrick, J.J.; Alexander, M.S. DOCK3 regulates normal skeletal muscle regeneration and glucose metabolism. FASEB J. 2023, 37, e23198. [Google Scholar] [CrossRef]
- Thompson, A.P.; Bitsina, C.; Gray, J.L.; von Delft, F.; Brennan, P.E. RHO to the DOCK for GDP disembarking: Structural insights into the DOCK GTPase nucleotide exchange factors. J. Biol. Chem. 2021, 296, 100521. [Google Scholar] [CrossRef] [PubMed]
- Ruan, W.; Pang, P.; Rao, Y. The SH2/SH3 Adaptor Protein Dock Interacts with the Ste20-like Kinase Misshapen in Controlling Growth Cone Motility. Neuron 1999, 24, 595–605. [Google Scholar] [CrossRef]
- Schlessinger, J. SH2/SH3 signaling proteins. Curr. Opin. Genet. Dev. 1994, 4, 25–30. [Google Scholar] [CrossRef]
- Rao, Y.; Zipursky, S.L. Domain requirements for the Dock adapter protein in growth- cone signaling. Proc. Natl. Acad. Sci. USA 1998, 95, 2077–2082. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, F.; Pan, Z.; Wang, W.; Wen, W. Solution structure of the SH3 domain of DOCK180. Proteins Struct. Funct. Bioinform. 2013, 81, 906–910. [Google Scholar] [CrossRef]
- Aitio, O.; Hellman, M.; Kazlauskas, A.; Vingadassalom, D.F.; Leong, J.M.; Saksela, K.; Permi, P. Recognition of tandem PxxP motifs as a unique Src homology 3-binding mode triggers pathogen-driven actin assembly. Proc. Natl. Acad. Sci. USA 2010, 107, 21743–21748. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Yang, J.; Jo, C.H.; Boland, A.; Zhang, Z.; McLaughlin, S.H.; Abu-Thuraia, A.; Killoran, R.C.; Smith, M.J.; Côté, J.-F.; et al. Structure of the DOCK2−ELMO1 complex provides insights into regulation of the auto-inhibited state. Nat. Commun. 2020, 11, 3464. [Google Scholar] [CrossRef] [PubMed]
- Hanawa-Suetsugu, K.; Kukimoto-Niino, M.; Mishima-Tsumagari, C.; Akasaka, R.; Ohsawa, N.; Sekine, S.-I.; Ito, T.; Tochio, N.; Koshiba, S.; Kigawa, T.; et al. Structural basis for mutual relief of the Rac guanine nucleotide exchange factor DOCK2 and its partner ELMO1 from their autoinhibited forms. Proc. Natl. Acad. Sci. USA 2012, 109, 3305–3310. [Google Scholar] [CrossRef] [PubMed]
- Biersmith, B.; Liu, Z.; Bauman, K.; Geisbrecht, E.R. The DOCK Protein Sponge Binds to ELMO and Functions in Drosophila Embryonic CNS Development. PLoS ONE 2011, 6, e16120. [Google Scholar] [CrossRef]
- Eguchi, K.; Yoshioka, Y.; Yoshida, H.; Morishita, K.; Miyata, S.; Hiai, H.; Yamaguchi, M. The Drosophila DOCK family protein sponge is involved in differentiation of R7 photoreceptor cells. Exp. Cell Res. 2013, 319, 2179–2195. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Peng, Y.; Wang, L.; Sun, X.; Wang, X.; Liang, C.; Yang, X.; Li, S.; Xu, J.; Ye, W.C.; et al. Autism-like social deficit generated by Dock4 deficiency is rescued by restoration of Rac1 activity and NMDA receptor function. Mol. Psychiatry 2019, 26, 1505–1519. [Google Scholar] [CrossRef]
- Zhou, C.; Licciulli, S.; Avila, J.L.; Cho, M.; Troutman, S.; Jiang, P.; Kossenkov, A.V.; Showe, L.C.; Liu, Q.; Vachani, A.; et al. The Rac1 splice form Rac1b promotes K-ras-induced lung tumorigenesis. Oncogene 2013, 32, 903–909. [Google Scholar] [CrossRef] [PubMed]
- Tejada-Simon, M.V. Modulation of actin dynamics by Rac1 to target cognitive function. J. Neurochem. 2015, 133, 767–779. [Google Scholar] [CrossRef]
- Jain, A.; Vale, R.D. RNA phase transitions in repeat expansion disorders. Nature 2017, 546, 243–247. [Google Scholar] [CrossRef]
- Bosco, E.E.; Mulloy, J.C.; Zheng, Y. Rac1 GTPase: A “Rac” of all trades. Cell. Mol. Life Sci. 2009, 66, 370–374. [Google Scholar] [CrossRef]
- Reijnders, M.R.F.; Ansor, N.M.; Kousi, M.; Yue, W.W.; Tan, P.L.; Clarkson, K.; Clayton-Smith, J.; Corning, K.; Jones, J.R.; Lam, W.W.K.; et al. RAC1 Missense Mutations in Developmental Disorders with Diverse Phenotypes. Am. J. Hum. Genet. 2017, 101, 466–477. [Google Scholar] [CrossRef]
- Sugihara, K.; Nakatsuji, N.; Nakamura, K.; Nakao, K.; Hashimoto, R.; Otani, H.; Sakagami, H.; Kondo, H.; Nozawa, S.; Aiba, A.; et al. Rac1 is required for the formation of three germ layers during gastrulation. Oncogene 1998, 17, 3427–3433. [Google Scholar] [CrossRef] [PubMed]
- Satoh, M.; Ogita, H.; Takeshita, K.; Mukai, Y.; Kwiatkowski, D.J.; Liao, J.K. Requirement of Rac1 in the development of cardiac hypertrophy. Proc. Natl. Acad. Sci. USA 2006, 103, 7432–7437. [Google Scholar] [CrossRef] [PubMed]
- Raun, S.H.; Ali, M.; Kjøbsted, R.; Møller, L.L.V.; Federspiel, M.A.; Richter, E.A.; Jensen, T.E.; Sylow, L. Rac1 muscle knockout exacerbates the detrimental effect of high-fat diet on insulin-stimulated muscle glucose uptake independently of Akt. J. Physiol. 2018, 596, 2283–2299. [Google Scholar] [CrossRef] [PubMed]
- Sylow, L.; Møller, L.L.; D’Hulst, G.; Schjerling, P.; Jensen, T.E.; Richter, E.A. Rac1 in Muscle Is Dispensable for Improved Insulin Action After Exercise in Mice. Endocrinology 2016, 157, 3009–3015. [Google Scholar] [CrossRef]
- Møller, L.L.V.; Ali, M.S.; Davey, J.; Raun, S.H.; Andersen, N.R.; Long, J.Z.; Qian, H.; Jeppesen, J.F.; Henriquez-Olguin, C.; Frank, E.; et al. The Rho guanine dissociation inhibitor α inhibits skeletal muscle Rac1 activity and insulin action. Proc. Natl. Acad. Sci. USA 2023, 120, e2211041120. [Google Scholar] [CrossRef]
- Kann, A.P.; Hung, M.; Wang, W.; Nguyen, J.; Gilbert, P.M.; Wu, Z.; Krauss, R.S. An injury-responsive Rac-to-Rho GTPase switch drives activation of muscle stem cells through rapid cytoskeletal remodeling. Cell Stem Cell 2022, 29, 933–947.e6. [Google Scholar] [CrossRef]
- Benson, C.A.; Olson, K.L.; Patwa, S.; Reimer, M.L.; Bangalore, L.; Hill, M.; Waxman, S.G.; Tan, A.M. Conditional RAC1 knockout in motor neurons restores H-reflex rate-dependent depression after spinal cord injury. Sci. Rep. 2021, 11, 7838. [Google Scholar] [CrossRef]
- Klockner, I.; Schutt, C.; Gerhardt, T.; Boettger, T.; Braun, T. Control of CRK-RAC1 activity by the miR-1/206/133 miRNA family is essential for neuromuscular junction function. Nat. Commun. 2022, 13, 3180. [Google Scholar] [CrossRef]
- Gumienny, T.L.; Brugnera, E.; Tosello-Trampont, A.C.; Kinchen, J.M.; Haney, L.B.; Nishiwaki, K.; Walk, S.F.; Nemergut, M.E.; Macara, I.G.; Francis, R.; et al. CED-12/ELMO, a novel member of the CrkII/Dock180/Rac pathway, is required for phagocytosis and cell migration. Cell 2001, 107, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Katoh, H.; Hiramoto, K.; Negishi, M. Activation of Rac1 by RhoG regulates cell migration. J. Cell Sci. 2006, 119, 56–65. [Google Scholar] [CrossRef]
- Laurin, M.; Fradet, N.; Blangy, A.; Hall, A.; Vuori, K.; Côté, J.F. The atypical Rac activator Dock180 (Dock1) regulates myoblast fusion in vivo. Proc. Natl. Acad. Sci. USA 2008, 105, 15446–15451. [Google Scholar] [CrossRef] [PubMed]
- Tran, V.; Nahlé, S.; Robert, A.; Desanlis, I.; Killoran, R.; Ehresmann, S.; Thibault, M.P.; Barford, D.; Ravichandran, K.S.; Sauvageau, M.; et al. Biasing the conformation of ELMO2 reveals that myoblast fusion can be exploited to improve muscle regeneration. Nat. Commun. 2022, 13, 7077. [Google Scholar] [CrossRef] [PubMed]
- Namekata, K.; Tsuji, N.; Guo, X.; Nishijima, E.; Honda, S.; Kitamura, Y.; Yamasaki, A.; Kishida, M.; Takeyama, J.; Ishikawa, H.; et al. Neuroprotection and axon regeneration by novel low-molecular-weight compounds through the modification of DOCK3 conformation. Cell Death Discov. 2023, 9, 166. [Google Scholar] [CrossRef]
- Banka, S.; Bennington, A.; Baker, M.J.; Rijckmans, E.; Clemente, G.D.; Ansor, N.M.; Sito, H.; Prasad, P.; Anyane-Yeboa, K.; Badalato, L.; et al. Activating RAC1 variants in the switch II region cause a developmental syndrome and alter neuronal morphology. Brain 2022, 145, 4232–4245. [Google Scholar] [CrossRef] [PubMed]
- Cetinkaya, A.; Xiong, J.R.; Vargel, I.; Kosemehmetoglu, K.; Canter, H.I.; Gerdan, O.F.; Longo, N.; Alzahrani, A.; Camps, M.P.; Taskiran, E.Z.; et al. Loss-of-function mutations in ELMO2 cause intraosseous vascular malformation by impeding RAC1 signaling. Am. J. Hum. Genet. 2016, 99, 299–317. [Google Scholar] [CrossRef]
- Clark, R.F.; Hutton, M.; Talbot, C.; Wragg, M.; Lendon, C.; Busfield, F.; Han, S.W.; Perez-Tur, J.; Adams, M.; Fuldner, R.; et al. The role of presenilin 1 in the genetics of Alzheimer’s disease. Cold Spring Harbor Symp. Quant. Biol. 1996, 61, 551–558. [Google Scholar]
- Laurin, M.; Côté, J.-F. Insights into the biological functions of Dock family guanine nucleotide exchange factors. Genes Dev. 2014, 28, 533–547. [Google Scholar] [CrossRef]
- Charleston, J.S.; Schnell, F.J.; Dworzak, J.; Donoghue, C.; Lewis, S.; Chen, L.; Young, G.D.; Milici, A.J.; Voss, J.; DeAlwis, U.; et al. Eteplirsen treatment for Duchenne muscular dystrophy. Neurology 2018, 90, e2146–e2154. [Google Scholar] [CrossRef]
- Mendell, J.R.; Goemans, N.; Lowes, L.P.; Alfano, L.N.; Berry, K.; Shao, J.; Kaye, E.M.; Mercuri, E.; for the Eteplirsen Study Group; Telethon Foundation DMD Italian Network. Longitudinal effect of eteplirsen versus historical control on ambulation in Duchenne muscular dystrophy. Ann. Neurol. 2016, 79, 257–271. [Google Scholar] [CrossRef]
- Iff, J.; Gerrits, C.; Zhong, Y.; Tuttle, E.; Birk, E.; Zheng, Y.; Paul, X.; Henricson, E.K.; McDonald, C.M.; Investigators, C.-D. Delays in pulmonary decline in eteplirsen-treated patients with Duchenne muscular dystrophy. Muscle Nerve 2022, 66, 262–269. [Google Scholar] [CrossRef] [PubMed]
- Vila-Perelló, M.; Muir, T.W. Biological Applications of Protein Splicing. Cell 2010, 143, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Stevens, A.J.; Sekar, G.; Shah, N.H.; Mostafavi, A.Z.; Cowburn, D.; Muir, T.W. A promiscuous split intein with expanded protein engineering applications. Proc. Natl. Acad. Sci. USA 2017, 114, 8538–8543. [Google Scholar] [CrossRef] [PubMed]
- Chadman, K.K.; Adayev, T.; Udayan, A.; Ahmed, R.; Dai, C.L.; Goodman, J.H.; Meeker, H.; Dolzhanskaya, N.; Velinov, M. Efficient Delivery of FMR1 across the Blood Brain Barrier Using AAVphp Construct in Adult FMR1 KO Mice Suggests the Feasibility of Gene Therapy for Fragile X Syndrome. Genes 2023, 14, 505. [Google Scholar] [CrossRef] [PubMed]
- Goertsen, D.; Flytzanis, N.C.; Goeden, N.; Chuapoco, M.R.; Cummins, A.; Chen, Y.; Fan, Y.; Zhang, Q.; Sharma, J.; Duan, Y.; et al. AAV capsid variants with brain-wide transgene expression and decreased liver targeting after intravenous delivery in mouse and marmoset. Nat. Neurosci. 2022, 25, 106–115. [Google Scholar] [CrossRef]
- Villiger, L.; Grisch-Chan, H.M.; Lindsay, H.; Ringnalda, F.; Pogliano, C.B.; Allegri, G.; Fingerhut, R.; Häberle, J.; Matos, J.; Robinson, M.D.; et al. Treatment of a metabolic liver disease by in vivo genome base editing in adult mice. Nat. Med. 2018, 24, 1519–1525. [Google Scholar] [CrossRef]
- Samani, A.; English, K.G.; Lopez, M.A.; Birch, C.L.; Brown, D.M.; Kaur, G.; Worthey, E.A.; Alexander, M.S. DOCKopathies: A systematic review of the clinical pathologies associated with human DOCK pathogenic variants. Hum. Mutat. 2022, 43, 1149–1161. [Google Scholar] [CrossRef]


{kind=link}
| Patient | DOCK3 Variant | Reference | Sex | Age at Evaluation | Birth History | Family History | Developmental Milestones | Growth (%) | Dysmorphic Features | Congenital Anomalies | Studies Prior to Diagnosis |
|---|---|---|---|---|---|---|---|---|---|---|---|
| P1 | del3:50789040- 51247265/c.382C>T | [10] | F | 12 years | Born at 42 weeks gestation | Similarly affected sibling | Severe Developmental Delay, walked at 5 years, unstable crouched, ataxic gait, non- verbal, and not toilet trained at 12y | WT = 25; Ht = 15; HC = 75 | Prominent chin, high arched palate, malocclusion, long fingers | None | Normal metabolic screen, EEG, BEAR, brain MRI |
| P2 | del3:50789040- 51247265/c.382C>T | [10] | M | 11 years | Born at 41 weeks gestation | Similarly affected sibling | Walked at 2.5 years, first word at 4 years, single words at TOE, unstable, ataxic gait | Wt = 40; Ht = 8; HC = 30 | Pointed chin, down slanting palpebral fissures, long face | None | None |
| P3 | homozygous del. 3:51,062,402–51,232,768 | [12] | M | 28 months | NR | Parents are first cousins | Started sitting at 14 months, walked at 22 months, unsteady gait, few specific words at TOE, Bayley score <50 (at TOE) | Wt = 4; Ht = 5; HC = 14 | Epicanthal folds, up-turned nasal tip, prominent cheeks | None | Brain MRI-dysmorphic Corpus Callosum, ECHO-normal |
| P4 | c.1038-2A>G:IVS12- 2A>G/c.3107_3110delACTT | [13] | M | 5 years | Born at 37 weeks gestation | Unremarkable | Started walking at 36 months, 5-10 single words at 5 | Wt ≥ 99; Ht = 66; HC = 85 | Broad forehead, deep set, hooded eyes | TE fistula with esophageal atresia, vertebral anomalies, rib anomalies, single kidney | Negative microarray, brain MRI- shallow sulci, hypoplastic white matter, spine MRI-syrinx, abnormal EEG |
| P5 | c.1175G>A/c.3887A>G | [13] | M | 5.5 years | Full term | Unremarkable | Was able to sit at 30 months, walked at 48 months, non-verbal, autism, unprovoked laughter, hypotonia | Wt = 50; Ht = 25; HC = 7 | Brachicephaly, plagiocephaly, prominent philtrum | Phimosis | Brain MRI-diminished white matter, hypoplastic CC, negative macroarray, UBE3A, MECP2, meth-Angelman |
| P6 | c.5020A>T/5020A>T | [13] | F | 3 years | born at 35 weeks gestation, feeding difficulties | NR | Walked at 18 months, and said the first word at 15 months, but then lost her speech, autism | Wt ≥ 99; Ht ≥ 99; HC ≥ 99 | Macrocephaly, frontal bossing | Spina bifida | Brain MRI-resolved Chiari malformation, negative CMA, PTEN, FXS |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alexander, M.S.; Velinov, M. DOCK3-Associated Neurodevelopmental Disorder—Clinical Features and Molecular Basis. Genes 2023, 14, 1940. https://doi.org/10.3390/genes14101940
Alexander MS, Velinov M. DOCK3-Associated Neurodevelopmental Disorder—Clinical Features and Molecular Basis. Genes. 2023; 14(10):1940. https://doi.org/10.3390/genes14101940
Chicago/Turabian StyleAlexander, Matthew S., and Milen Velinov. 2023. "DOCK3-Associated Neurodevelopmental Disorder—Clinical Features and Molecular Basis" Genes 14, no. 10: 1940. https://doi.org/10.3390/genes14101940