
Changes in Hox Gene Chromatin Organization during Odontogenic Lineage Specification

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Isolation and Culture of Human Odontogenic Progenitors
2.2. Mineralization Induction of DF, AB, and CNCC
2.3. Chip-on-Chip Analysis
2.4. Sample Preparation for ChIP-Chip, Whole Genome Amplification, Dual-Color Labeling, and Array Scanning
2.5. Raw Data Processing and Gene Annotation
2.6. Data Processing for Visualizing ChIP-Chip and ChIP-Seq Enrichment
2.7. Chromatin Immunoprecipitation Analysis
2.8. Cdx4 Plasmid Constructs and Transfections
2.9. RNA Extraction and RT PCR
2.10. Immunohistochemistry
2.11. Mouse Tooth Organ Culture and EZH2 Inhibitor Treatment
2.12. Statistical Analysis
3. Results
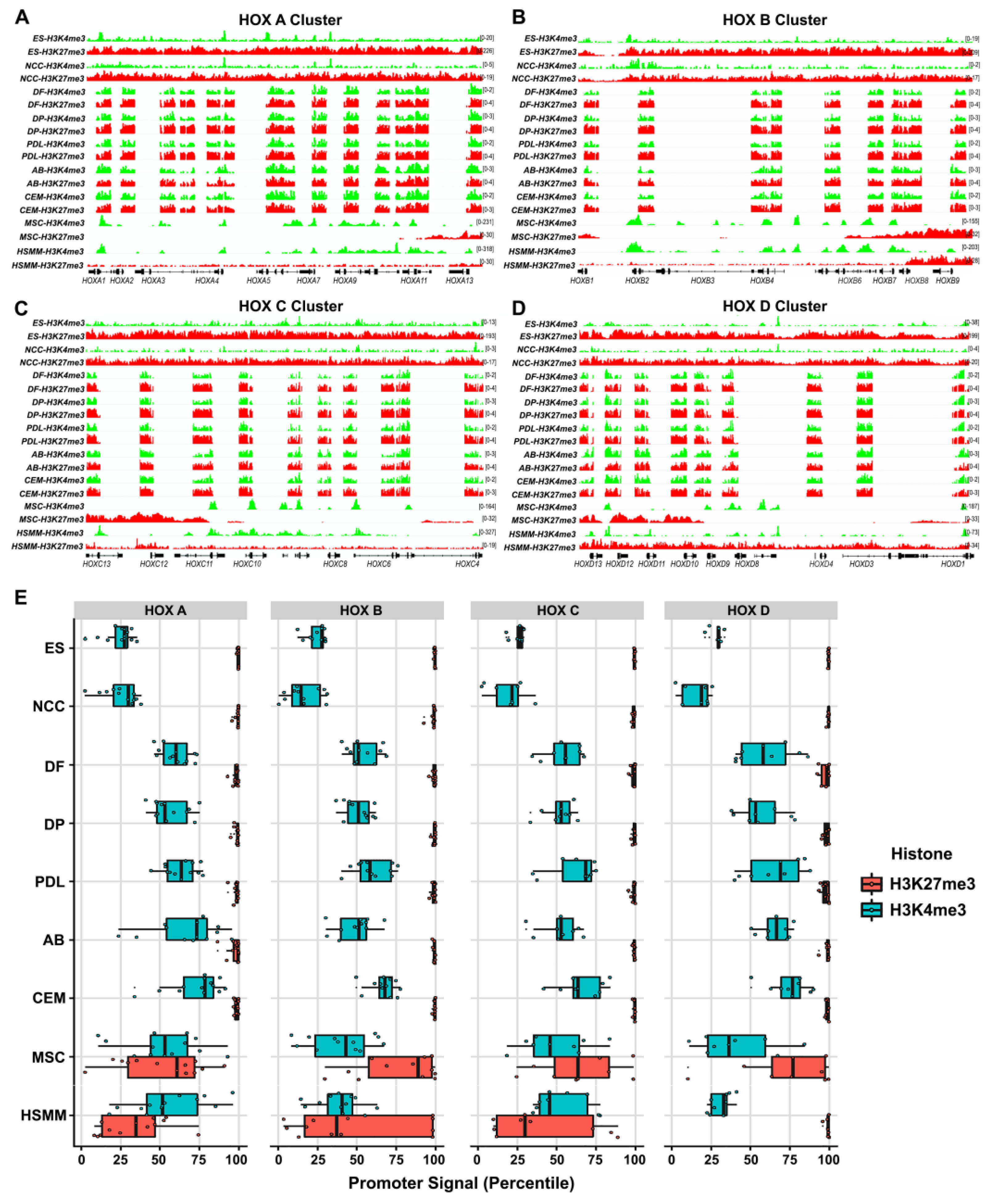
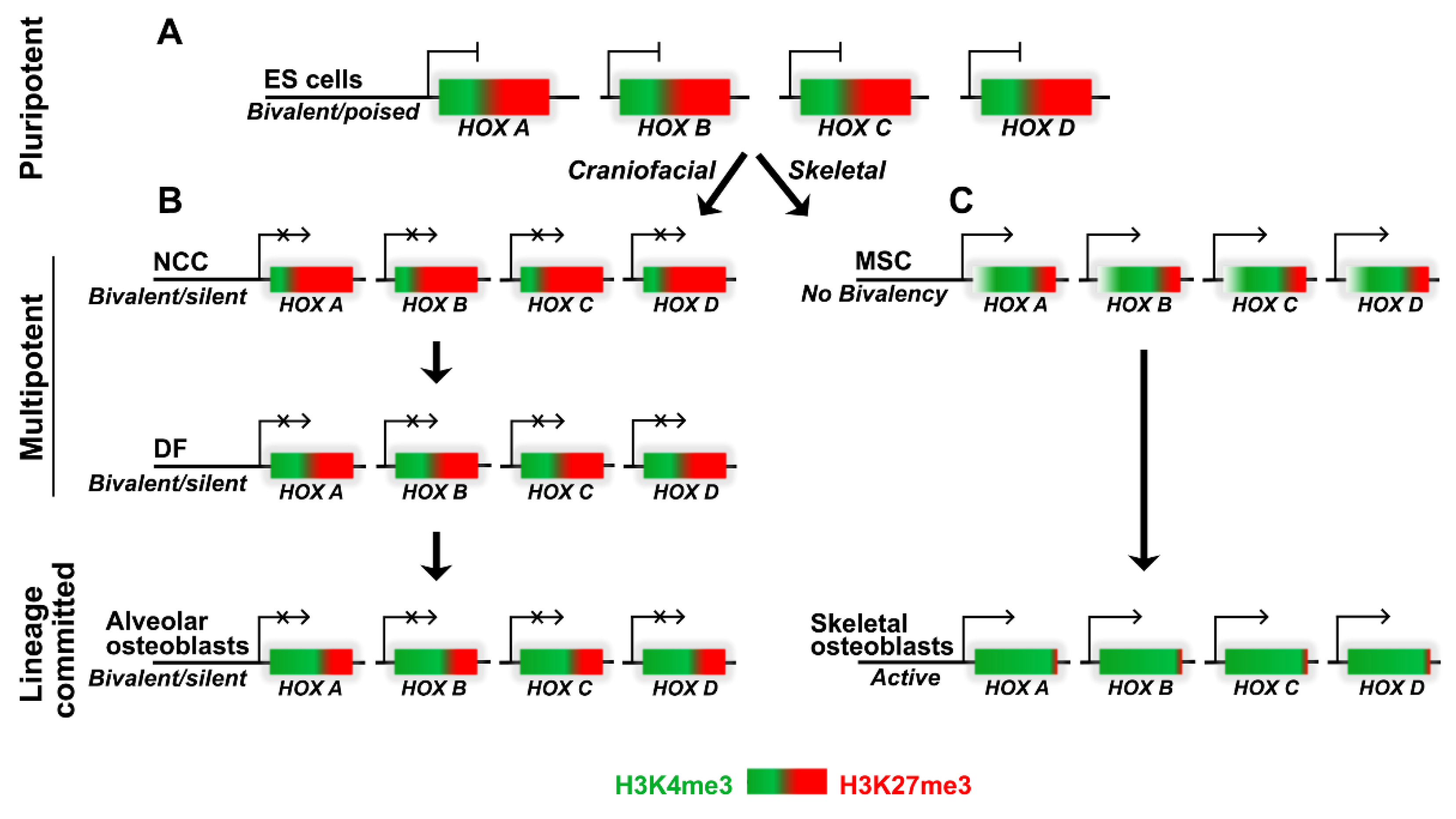
3.1. Repressive Histone Modifications Marked by H3K27me3 at HOX Promoters Distinguish Craniofacial Mesenchymal Progenitors from Trunk Lineages
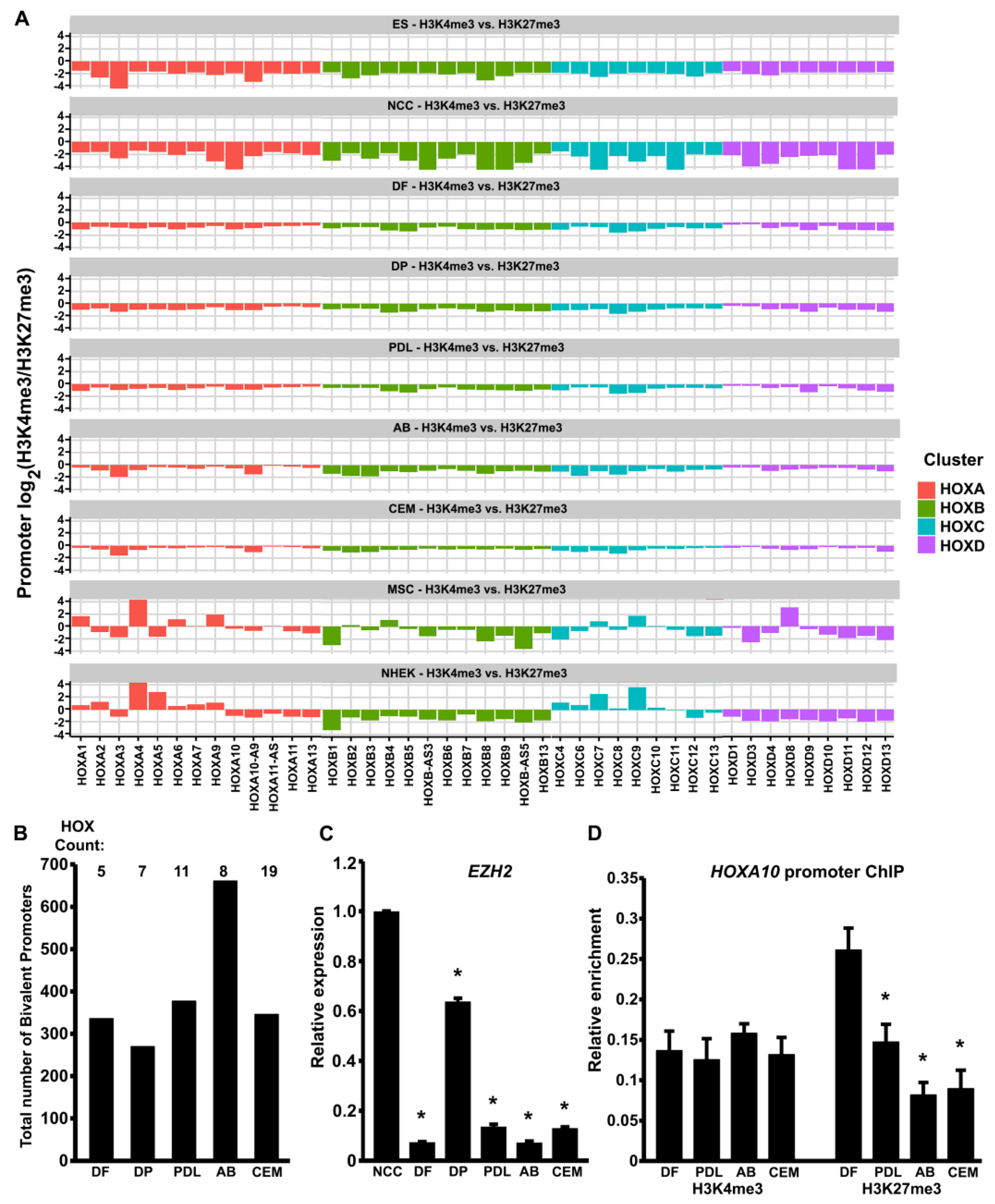
3.2. HOX Gene Promoters in Odontogenic Progenitors Were Characterized by a Bivalent Histone Methylation Signature
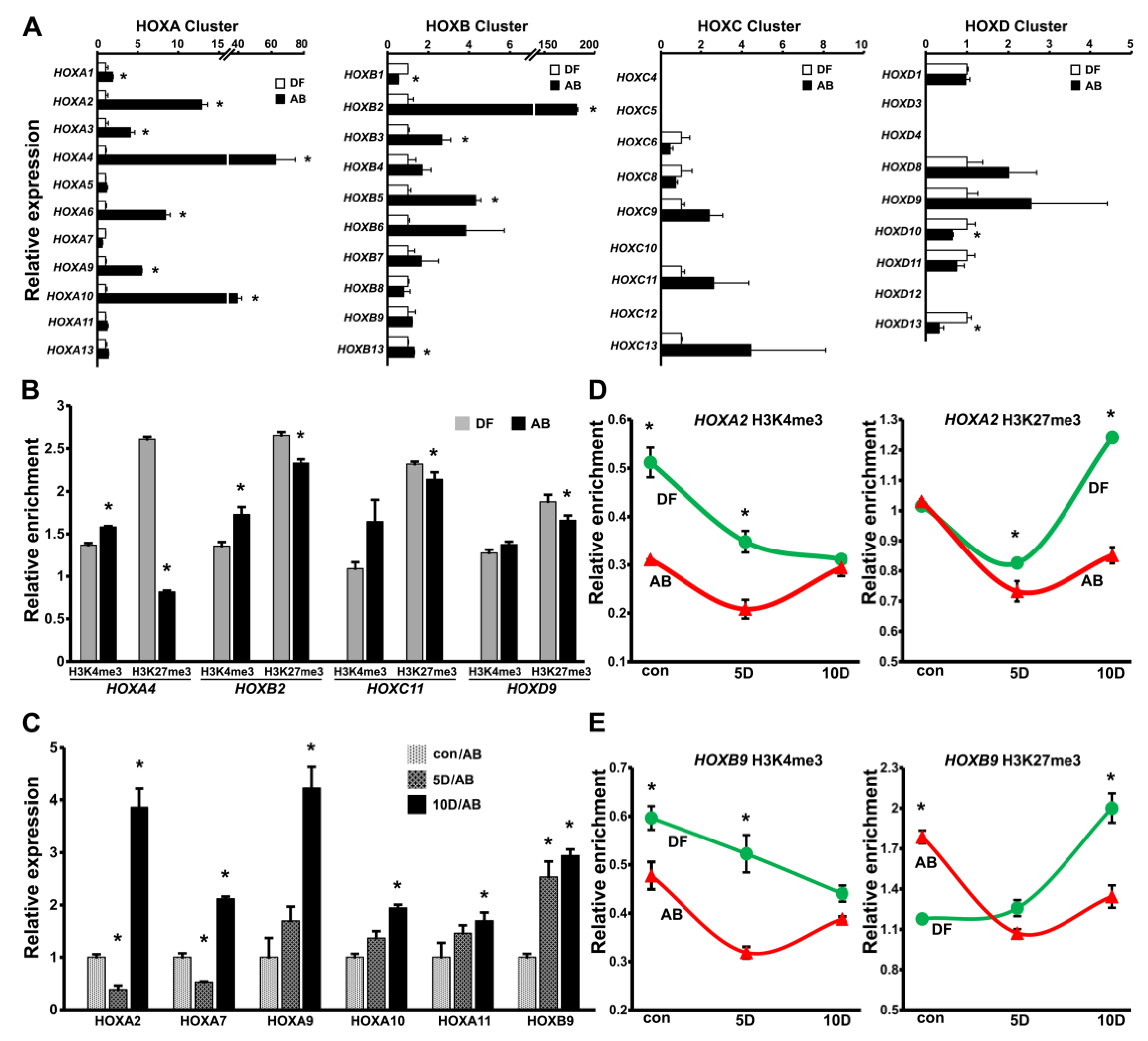
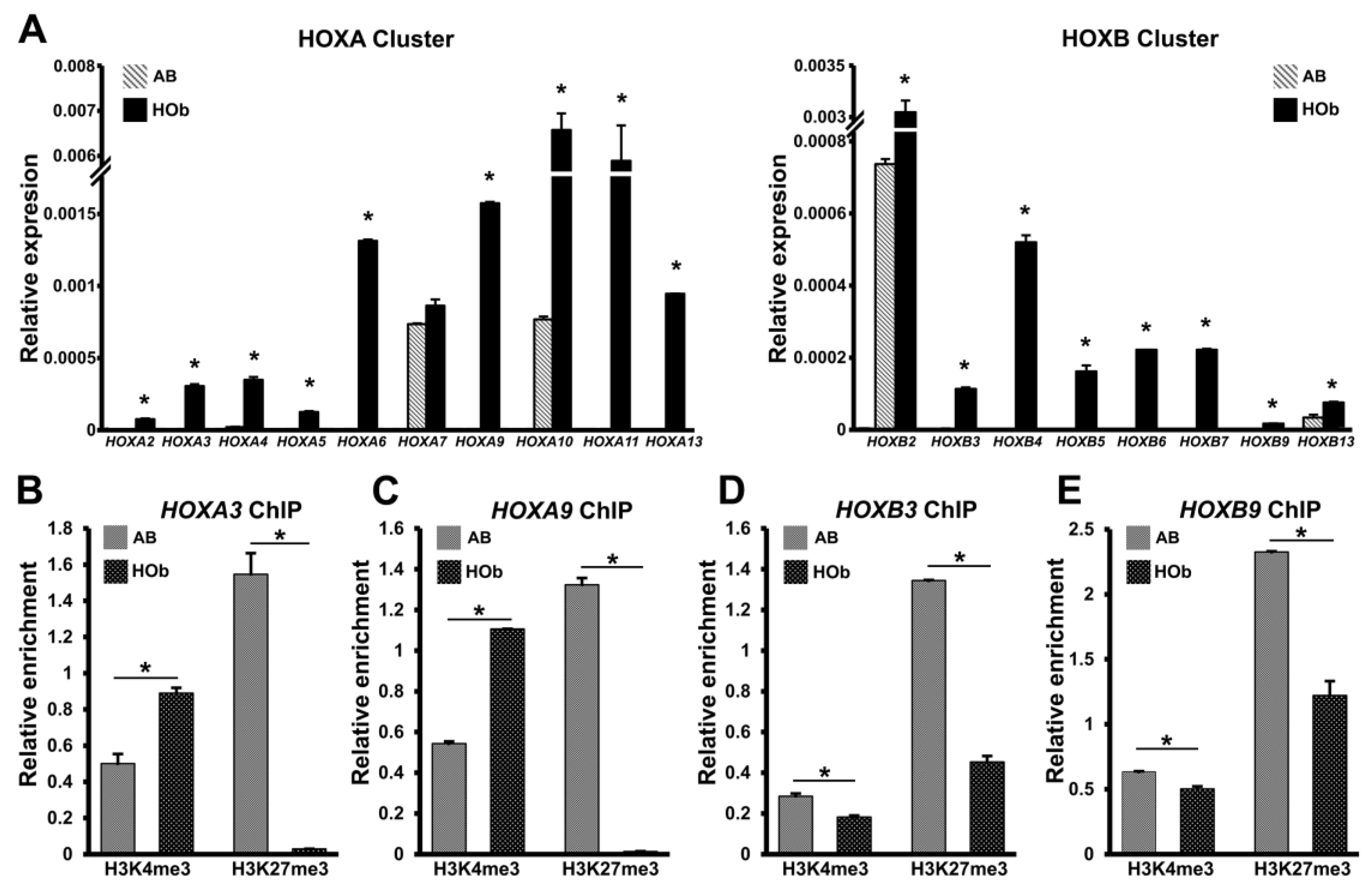
3.3. Higher Levels of HOX Gene Expression and Reduced Levels of H3K27me3 Histone Marks in AB Cells Versus DF Cells
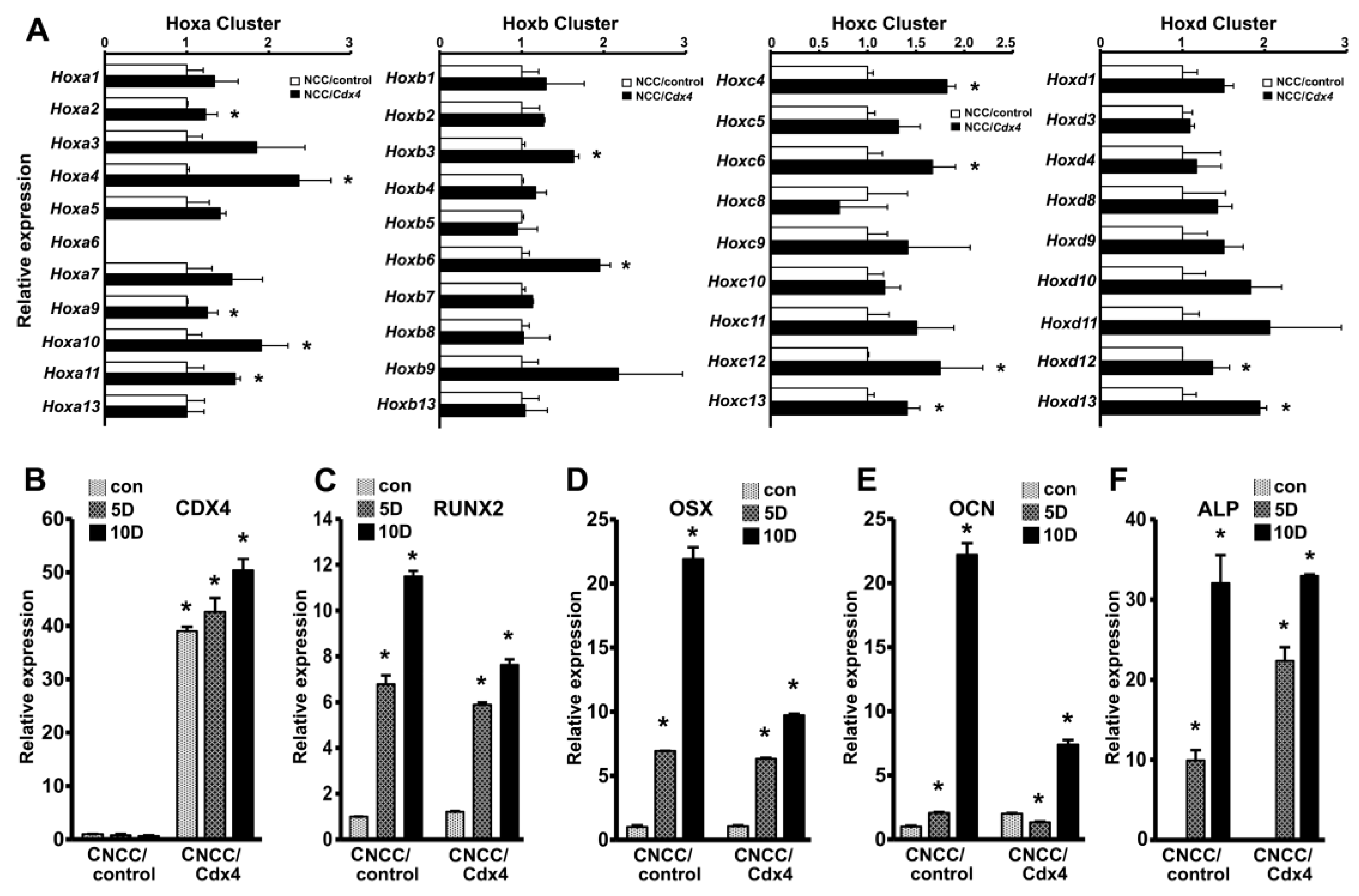
3.4. Cdx4 Overexpression Induces HOX Gene Upregulation and Decreased Mineralization Marker Gene Expression in Mouse Cranial Neural Crest Cells
3.5. Elevated HOX Gene Expression and Decreased H3K27me3 Marks on HOX Promoters of Human Skeletal Bone Osteoblasts Compared to Craniofacial Alveolar Bone Progenitors
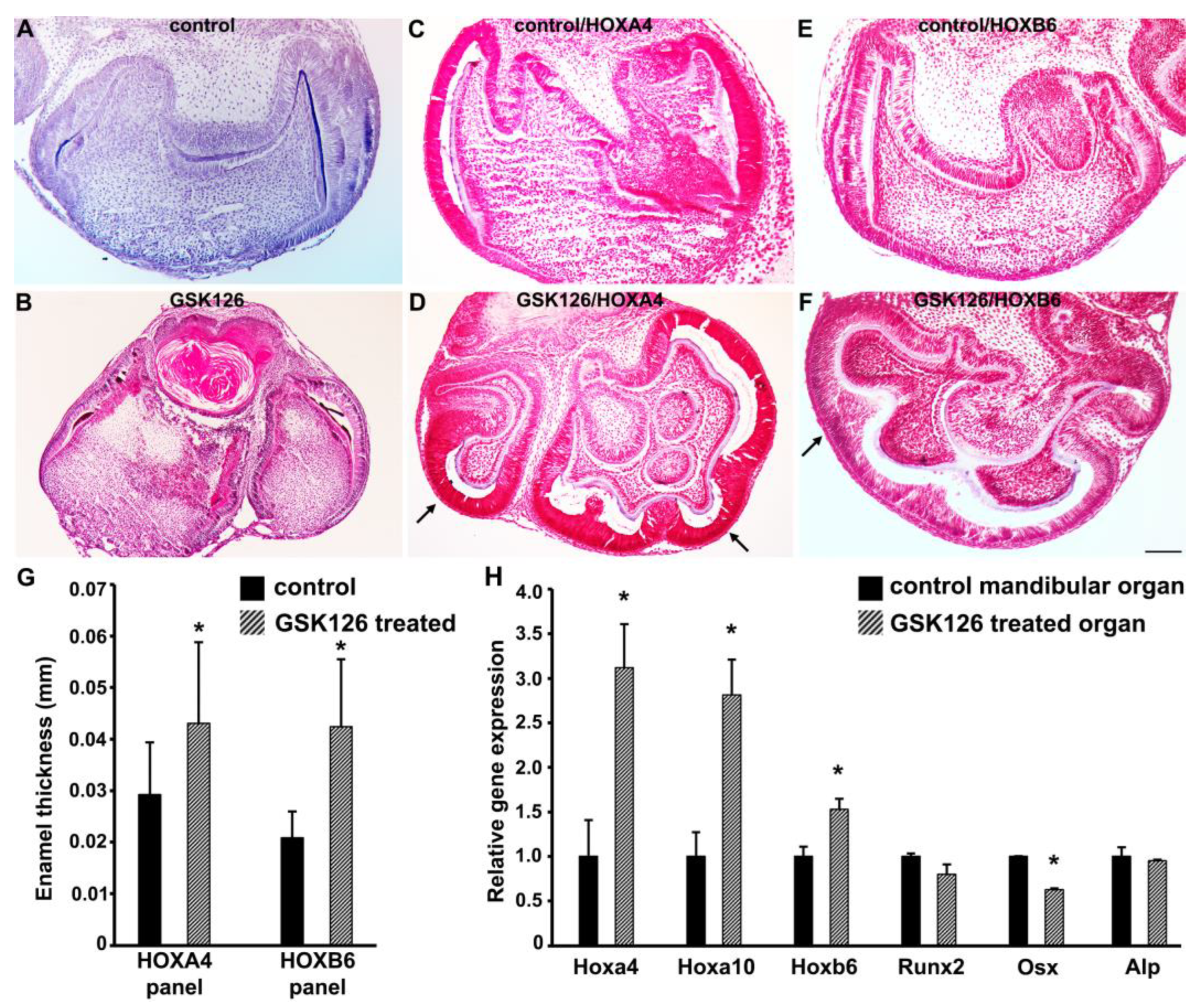
3.6. Small Molecule-Mediated EZH2 Inhibition Leads to Increased Patterning and Supernumerary Cusp Formation in Mouse Tooth Organs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gans, C.; Northcutt, R.G. Neural Crest and the Origin of Vertebrates: A New Head. Science 1983, 220, 268–273. [Google Scholar] [CrossRef] [PubMed]
- Northcutt, R.G.; Gans, C. The Genesis of Neural Crest and Epidermal Placodes: A Reinterpretation of Vertebrate Origins. Q. Rev. Biol. 1983, 58, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Trainor, P.A.; Melton, K.R.; Manzanares, M. Origins and Plasticity of Neural Crest Cells and Their Roles in Jaw and Craniofacial Evolution. Int. J. Dev. Biol. 2003, 47, 541–553. [Google Scholar] [PubMed]
- Creuzet, S.E.; Martinez, S.; Le Douarin, N.M. The Cephalic Neural Crest Exerts a Critical Effect on Forebrain and Midbrain Development. Proc. Natl. Acad. Sci. USA 2006, 103, 14033–14038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Douarin, N.M.; Brito, J.M.; Creuzet, S. Role of the Neural Crest in Face and Brain Development. Brain Res. Rev. 2007, 55, 237–247. [Google Scholar] [CrossRef] [Green Version]
- Bronner-Fraser, M.; García-Castro, M. Chapter 4 Manipulations of Neural Crest Cells or Their Migratory Pathways. In Methods in Cell Biology; Elsevier: Amsterdam, The Netherlands, 2008; Volume 87, pp. 75–96. ISBN 9780125641746. [Google Scholar]
- Kanzler, B.; Kuschert, S.J.; Liu, Y.H.; Mallo, M. Hoxa-2 Restricts the Chondrogenic Domain and Inhibits Bone Formation during Development of the Branchial Area. Development 1998, 125, 2587–2597. [Google Scholar] [CrossRef]
- Minoux, M.; Rijli, F.M. Molecular Mechanisms of Cranial Neural Crest Cell Migration and Patterning in Craniofacial Development. Development 2010, 137, 2605–2621. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, D.; Varum, S.; Zemke, M.; Schöler, A.; Baggiolini, A.; Draganova, K.; Koseki, H.; Schübeler, D.; Sommer, L. Ezh2 Is Required for Neural Crest-Derived Cartilage and Bone Formation. Development 2014, 141, 867–877. [Google Scholar] [CrossRef] [Green Version]
- Kitazawa, T.; Minoux, M.; Ducret, S.; Rijli, F.M. Different Ectopic Hoxa2 Expression Levels in Mouse Cranial Neural Crest Cells Result in Distinct Craniofacial Anomalies and Homeotic Phenotypes. JDB 2022, 10, 9. [Google Scholar] [CrossRef]
- McGinnis, W.; Levine, M.S.; Hafen, E.; Kuroiwa, A.; Gehring, W.J. A Conserved DNA Sequence in Homoeotic Genes of the Drosophila Antennapedia and Bithorax Complexes. Nature 1984, 308, 428–433. [Google Scholar] [CrossRef]
- Scott, M.P.; Weiner, A.J. Structural Relationships among Genes That Control Development: Sequence Homology between the Antennapedia, Ultrabithorax, and Fushi Tarazu Loci of Drosophila. Proc. Natl. Acad. Sci. USA 1984, 81, 4115–4119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGinnis, W.; Krumlauf, R. Homeobox Genes and Axial Patterning. Cell 1992, 68, 283–302. [Google Scholar] [CrossRef] [PubMed]
- Mallo, M.; Wellik, D.M.; Deschamps, J. Hox Genes and Regional Patterning of the Vertebrate Body Plan. Dev. Biol. 2010, 344, 7–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kessel, M.; Gruss, P. Murine Developmental Control Genes. Science 1990, 249, 374–379. [Google Scholar] [CrossRef] [Green Version]
- Duboule, D.; Morata, G. Colinearity and Functional Hierarchy among Genes of the Homeotic Complexes. Trends Genet. 1994, 10, 358–364. [Google Scholar] [CrossRef]
- Krumlauf, R. Hox Genes and Pattern Formation in the Branchial Region of the Vertebrate Head. Trends Genet. 1993, 9, 106–112. [Google Scholar] [CrossRef]
- Manley, N.R.; Capecchi, M.R. HoxGroup 3 Paralogs Regulate the Development and Migration of the Thymus, Thyroid, and Parathyroid Glands. Dev. Biol. 1998, 195, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Mallo, M.; Vinagre, T.; Carapuo, M. The Road to the Vertebral Formula. Int. J. Dev. Biol. 2009, 53, 1469–1481. [Google Scholar] [CrossRef] [Green Version]
- Wellik, D.M. Chapter 9 Hox Genes and Vertebrate Axial Pattern. In Current Topics in Developmental Biology; Elsevier: Amsterdam, The Netherlands, 2009; Volume 88, pp. 257–278. ISBN 9780123745293. [Google Scholar]
- Catón, J.; Tucker, A.S. Current Knowledge of Tooth Development: Patterning and Mineralization of the Murine Dentition. J. Anat. 2009, 214, 502–515. [Google Scholar] [CrossRef]
- Thomas, B.L.; Tucker, A.S.; Qui, M.; Ferguson, C.A.; Hardcastle, Z.; Rubenstein, J.L.; Sharpe, P.T. Role of Dlx-1 and Dlx-2 Genes in Patterning of the Murine Dentition. Development 1997, 124, 4811–4818. [Google Scholar] [CrossRef]
- Tucker, A.S.; Matthews, K.L.; Sharpe, P.T. Transformation of Tooth Type Induced by Inhibition of BMP Signaling. Science 1998, 282, 1136–1138. [Google Scholar] [CrossRef] [PubMed]
- Tucker, A.S.; Yamada, G.; Grigoriou, M.; Pachnis, V.; Sharpe, P.T. Fgf-8 Determines Rostral-Caudal Polarity in the First Branchial Arch. Development 1999, 126, 51–61. [Google Scholar] [CrossRef]
- Mitsiadis, T.A.; Drouin, J. Deletion of the Pitx1 Genomic Locus Affects Mandibular Tooth Morphogenesis and Expression of the Barx1 and Tbx1 Genes. Dev. Biol. 2008, 313, 887–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atkinson, S.P.; Koch, C.M.; Clelland, G.K.; Willcox, S.; Fowler, J.C.; Stewart, R.; Lako, M.; Dunham, I.; Armstrong, L. Epigenetic Marking Prepares the Human HOXA Cluster for Activation During Differentiation of Pluripotent Cells. Stem Cells 2008, 26, 1174–1185. [Google Scholar] [CrossRef] [PubMed]
- Gan, Q.; Yoshida, T.; McDonald, O.G.; Owens, G.K. Concise Review: Epigenetic Mechanisms Contribute to Pluripotency and Cell Lineage Determination of Embryonic Stem Cells. Stem Cells 2007, 25, 2–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barber, B.A.; Rastegar, M. Epigenetic Control of Hox Genes during Neurogenesis, Development, and Disease. Ann. Anat.-Anat. Anz. 2010, 192, 261–274. [Google Scholar] [CrossRef]
- Cui, K.; Zang, C.; Roh, T.-Y.; Schones, D.E.; Childs, R.W.; Peng, W.; Zhao, K. Chromatin Signatures in Multipotent Human Hematopoietic Stem Cells Indicate the Fate of Bivalent Genes during Differentiation. Cell Stem Cell 2009, 4, 80–93. [Google Scholar] [CrossRef] [Green Version]
- Meissner, A.; Mikkelsen, T.S.; Gu, H.; Wernig, M.; Hanna, J.; Sivachenko, A.; Zhang, X.; Bernstein, B.E.; Nusbaum, C.; Jaffe, D.B.; et al. Genome-Scale DNA Methylation Maps of Pluripotent and Differentiated Cells. Nature 2008, 454, 766–770. [Google Scholar] [CrossRef] [Green Version]
- Wei, G.; Wei, L.; Zhu, J.; Zang, C.; Hu-Li, J.; Yao, Z.; Cui, K.; Kanno, Y.; Roh, T.-Y.; Watford, W.T.; et al. Global Mapping of H3K4me3 and H3K27me3 Reveals Specificity and Plasticity in Lineage Fate Determination of Differentiating CD4+ T Cells. Immunity 2009, 30, 155–167. [Google Scholar] [CrossRef] [Green Version]
- Ringrose, L.; Paro, R. Polycomb/Trithorax Response Elements and Epigenetic Memory of Cell Identity. Development 2007, 134, 223–232. [Google Scholar] [CrossRef]
- Kanduri, M.; Sander, B.; Ntoufa, S.; Papakonstantinou, N.; Sutton, L.-A.; Stamatopoulos, K.; Kanduri, C.; Rosenquist, R. A Key Role for EZH2 in Epigenetic Silencing of HOX Genes in Mantle Cell Lymphoma. Epigenetics 2013, 8, 1280–1288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, F.; Liu, L.; Chang, C.-K.; He, Q.; Wu, L.-Y.; Zhang, Z.; Shi, W.-H.; Guo, J.; Zhu, Y.; Zhao, Y.-S.; et al. Genomic Loss of EZH2 Leads to Epigenetic Modifications and Overexpression of the HOX Gene Clusters in Myelodysplastic Syndrome. Oncotarget 2016, 7, 8119–8130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gopinathan, G.; Kolokythas, A.; Luan, X.; Diekwisch, T.G.H. Epigenetic Marks Define the Lineage and Differentiation Potential of Two Distinct Neural Crest-Derived Intermediate Odontogenic Progenitor Populations. Stem Cells Dev. 2013, 22, 1763–1778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative Genomics Viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondrashov, N.; Pusic, A.; Stumpf, C.R.; Shimizu, K.; Hsieh, A.C.; Xue, S.; Ishijima, J.; Shiroishi, T.; Barna, M. Ribosome-Mediated Specificity in Hox MRNA Translation and Vertebrate Tissue Patterning. Cell 2011, 145, 383–397. [Google Scholar] [CrossRef] [Green Version]
- Dickson, G.J.; Lappin, T.R.; Thompson, A. Complete Array of HOX Gene Expression by RQ-PCR. In Leukemia; Eric So, C.W., Ed.; Humana Press: Totowa, NJ, USA, 2009; Volume 538, pp. 369–393. ISBN 9781588299895. [Google Scholar]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Couly, G.F.; Coltey, P.M.; Le Douarin, N.M. The Triple Origin of Skull in Higher Vertebrates: A Study in Quail-Chick Chimeras. Development 1993, 117, 409–429. [Google Scholar] [CrossRef]
- Grapin-Botton, A.; Bonnin, M.A.; McNaughton, L.A.; Krumlauf, R.; Le Douarin, N.M. Plasticity of Transposed Rhombomeres: Hox Gene Induction Is Correlated with Phenotypic Modifications. Development 1995, 121, 2707–2721. [Google Scholar] [CrossRef]
- Douarin, N.L.; Kalcheim, C. The Neural Crest, 2nd ed.; Cambridge University Press: New York, NY, USA, 1999. [Google Scholar]
- Creuzet, S.; Couly, G.; Vincent, C.; Le Douarin, N.M. Negative Effect of Hox Gene Expression on the Development of the Neural Crest-Derived Facial Skeleton. Development 2002, 129, 4301–4313. [Google Scholar] [CrossRef]
- Azuara, V.; Perry, P.; Sauer, S.; Spivakov, M.; Jørgensen, H.F.; John, R.M.; Gouti, M.; Casanova, M.; Warnes, G.; Merkenschlager, M.; et al. Chromatin Signatures of Pluripotent Cell Lines. Nat. Cell Biol. 2006, 8, 532–538. [Google Scholar] [CrossRef]
- Bernstein, B.E.; Mikkelsen, T.S.; Xie, X.; Kamal, M.; Huebert, D.J.; Cuff, J.; Fry, B.; Meissner, A.; Wernig, M.; Plath, K.; et al. A Bivalent Chromatin Structure Marks Key Developmental Genes in Embryonic Stem Cells. Cell 2006, 125, 315–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldberg, A.D.; Allis, C.D.; Bernstein, E. Epigenetics: A Landscape Takes Shape. Cell 2007, 128, 635–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soshnikova, N.; Duboule, D. Epigenetic Regulation OfHox Gene Activation: The Waltz of Methyls. Bioessays 2008, 30, 199–202. [Google Scholar] [CrossRef]
- Morsczeck, C.; Schmalz, G.; Reichert, T.E.; Völlner, F.; Galler, K.; Driemel, O. Somatic Stem Cells for Regenerative Dentistry. Clin. Oral Investig. 2008, 12, 113–118. [Google Scholar] [CrossRef]
- Saugspier, M.; Felthaus, O.; Viale-Bouroncle, S.; Driemel, O.; Reichert, T.E.; Schmalz, G.; Morsczeck, C. The Differentiation and Gene Expression Profile of Human Dental Follicle Cells. Stem Cells Dev. 2010, 19, 707–717. [Google Scholar] [CrossRef]
- Charite, J.; de Graaff, W.; Consten, D.; Reijnen, M.J.; Korving, J.; Deschamps, J. Transducing Positional Information to the Hox Genes: Critical Interaction of Cdx Gene Products with Position-Sensitive Regulatory Elements. Development 1998, 125, 4349–4358. [Google Scholar] [CrossRef]
- Chai, Y.; Jiang, X.; Ito, Y.; Bringas, P.; Han, J.; Rowitch, D.H.; Soriano, P.; McMahon, A.P.; Sucov, H.M. Fate of the Mammalian Cranial Neural Crest during Tooth and Mandibular Morphogenesis. Development 2000, 127, 1671–1679. [Google Scholar] [CrossRef]
- Leucht, P.; Kim, J.-B.; Amasha, R.; James, A.W.; Girod, S.; Helms, J.A. Embryonic Origin and Hox Status Determine Progenitor Cell Fate during Adult Bone Regeneration. Development 2008, 135, 2845–2854. [Google Scholar] [CrossRef] [Green Version]
- Tan, J.; Yan, Y.; Wang, X.; Jiang, Y.; Xu, H.E. EZH2: Biology, Disease, and Structure-Based Drug Discovery. Acta Pharm. Sin. 2014, 35, 161–174. [Google Scholar] [CrossRef] [Green Version]
- Wiles, E.T.; Selker, E.U. H3K27 Methylation: A Promiscuous Repressive Chromatin Mark. Curr. Opin. Genet. Dev. 2017, 43, 31–37. [Google Scholar] [CrossRef]
- Cai, C.; Peng, X.; Zhang, Y. Downregulation of Cell Division Cycle-Associated Protein 7 (CDCA7) Suppresses Cell Proliferation, Arrests Cell Cycle of Ovarian Cancer, and Restrains Angiogenesis by Modulating Enhancer of Zeste Homolog 2 (EZH2) Expression. Bioengineered 2021, 12, 7007–7019. [Google Scholar] [CrossRef]
- Dangaria, S.J.; Ito, Y.; Luan, X.; Diekwisch, T.G.H. Differentiation of Neural-Crest-Derived Intermediate Pluripotent Progenitors into Committed Periodontal Populations Involves Unique Molecular Signature Changes, Cohort Shifts, and Epigenetic Modifications. Stem Cells Dev. 2011, 20, 39–52. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Langohr, I.M.; Faisal, M.; McNulty, M.; Thorn, C.; Kim, J. Ablation of Ezh2 in Neural Crest Cells Leads to Aberrant Enteric Nervous System Development in Mice. PLoS ONE 2018, 13, e0203391. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Wang, L.; Pei, P.; Li, X.; Wu, J.; Qiu, Z.; Zhang, J.; Ao, R.; Wang, S.; Zhang, T.; et al. Reduced H3K27me3 Leads to Abnormal Hox Gene Expression in Neural Tube Defects. Epigenet. Chromatin 2019, 12, 76. [Google Scholar] [CrossRef] [Green Version]
- Francis, M.; Gopinathan, G.; Foyle, D.; Fallah, P.; Gonzalez, M.; Luan, X.; Diekwisch, T.G.H. Histone Methylation: Achilles Heel and Powerful Mediator of Periodontal Homeostasis. J. Dent. Res. 2020, 99, 1332–1340. [Google Scholar] [CrossRef]
- Li, F.; Wan, M.; Zhang, B.; Peng, Y.; Zhou, Y.; Pi, C.; Xu, X.; Ye, L.; Zhou, X.; Zheng, L. Bivalent Histone Modifications and Development. CSCR 2018, 13, 83–90. [Google Scholar] [CrossRef]
- Pan, G.; Tian, S.; Nie, J.; Yang, C.; Ruotti, V.; Wei, H.; Jonsdottir, G.A.; Stewart, R.; Thomson, J.A. Whole-Genome Analysis of Histone H3 Lysine 4 and Lysine 27 Methylation in Human Embryonic Stem Cells. Cell Stem Cell 2007, 1, 299–312. [Google Scholar] [CrossRef] [Green Version]
- Minoux, M.; Holwerda, S.; Vitobello, A.; Kitazawa, T.; Kohler, H.; Stadler, M.B.; Rijli, F.M. Gene Bivalency at Polycomb Domains Regulates Cranial Neural Crest Positional Identity. Science 2017, 355, eaal2913. [Google Scholar] [CrossRef]
- Mikkelsen, T.S.; Ku, M.; Jaffe, D.B.; Issac, B.; Lieberman, E.; Giannoukos, G.; Alvarez, P.; Brockman, W.; Kim, T.-K.; Koche, R.P.; et al. Genome-Wide Maps of Chromatin State in Pluripotent and Lineage-Committed Cells. Nature 2007, 448, 553–560. [Google Scholar] [CrossRef] [Green Version]
- Voigt, P.; Tee, W.-W.; Reinberg, D. A Double Take on Bivalent Promoters. Genes Dev. 2013, 27, 1318–1338. [Google Scholar] [CrossRef]
- Stelnicki, E.J.; Kömüves, L.G.; Kwong, A.O.; Holmes, D.; Klein, P.; Rozenfeld, S.; Jeffrey Lawrence, H.; Scott Adzick, N.; Harrison, M.; Largman, C. HOX Homeobox Genes Exhibit Spatial and Temporal Changes in Expression during Human Skin Development. J. Investig. Dermatol. 1998, 110, 110–115. [Google Scholar] [CrossRef] [Green Version]
- Cordero, D.R.; Brugmann, S.; Chu, Y.; Bajpai, R.; Jame, M.; Helms, J.A. Cranial Neural Crest Cells on the Move: Their Roles in Craniofacial Development. Am. J. Med. Genet. 2011, 155, 270–279. [Google Scholar] [CrossRef] [Green Version]
- Helms, J.A.; Schneider, R.A. Cranial Skeletal Biology. Nature 2003, 423, 326–331. [Google Scholar] [CrossRef]
- Whiting, J. Craniofacial Abnormalities Induced by the Ectopic Expression of Homeobox Genes. Mutat. Res./Fundam. Mol. Mech. Mutagen. 1997, 396, 97–112. [Google Scholar] [CrossRef]
- Lufkin, T.; Mark, M.; Hart, C.P.; Dollé, P.; LeMeur, M.; Chambon, P. Homeotic Transformation of the Occipital Bones of the Skull by Ectopic Expression of a Homeobox Gene. Nature 1992, 359, 835–841. [Google Scholar] [CrossRef]
- Chisaka, O.; Musci, T.S.; Capecchi, M.R. Developmental Defects of the Ear, Cranial Nerves and Hindbrain Resulting from Targeted Disruption of the Mouse Homeobox GeneHox-#150;1.6. Nature 1992, 355, 516–520. [Google Scholar] [CrossRef]
- Mark, M.; Lufkin, T.; Vonesch, J.L.; Ruberte, E.; Olivo, J.C.; Dolle, P.; Gorry, P.; Lumsden, A.; Chambon, P. Two Rhombomeres Are Altered in Hoxa-1 Mutant Mice. Development 1993, 119, 319–338. [Google Scholar] [CrossRef]
- Goddard, J.M.; Rossel, M.; Manley, N.R.; Capecchi, M.R. Mice with Targeted Disruption of Hoxb-1 Fail to Form the Motor Nucleus of the VIIth Nerve. Development 1996, 122, 3217–3228. [Google Scholar] [CrossRef]
- Studer, M.; Lumsden, A.; Ariza-McNaughton, L.; Bradley, A.; Krumlauf, R. Altered Segmental Identity and Abnormal Migration of Motor Neurons in Mice Lacking Hoxb-1. Nature 1996, 384, 630–634. [Google Scholar] [CrossRef]
- Duboule, D. The Rise and Fall of Hox Gene Clusters. Development 2007, 134, 2549–2560. [Google Scholar] [CrossRef] [PubMed]
- Alexander, T.; Nolte, C.; Krumlauf, R. Hox Genes and Segmentation of the Hindbrain and Axial Skeleton. Annu. Rev. Cell Dev. Biol. 2009, 25, 431–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denans, N.; Iimura, T.; Pourquié, O. Hox Genes Control Vertebrate Body Elongation by Collinear Wnt Repression. eLife 2015, 4, e04379. [Google Scholar] [CrossRef] [PubMed]
- Parker, H.J.; Pushel, I.; Krumlauf, R. Coupling the Roles of Hox Genes to Regulatory Networks Patterning Cranial Neural Crest. Dev. Biol. 2018, 444, S67–S78. [Google Scholar] [CrossRef] [PubMed]
- Hall, B.K.; Hall, B.K. The Neural Crest and Neural Crest Cells in Vertebrate Development and Evolution, 2nd ed.; Springer: New York, NY, USA, 2009; ISBN 9780387098456. [Google Scholar]
- York, J.R.; McCauley, D.W. The Origin and Evolution of Vertebrate Neural Crest Cells. Open Biol. 2020, 10, 190285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simões-Costa, M.; Bronner, M.E. Establishing Neural Crest Identity: A Gene Regulatory Recipe. Development 2015, 142, 242–257. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gopinathan, G.; Zhang, X.; Luan, X.; Diekwisch, T.G.H. Changes in Hox Gene Chromatin Organization during Odontogenic Lineage Specification. Genes 2023, 14, 198. https://doi.org/10.3390/genes14010198
Gopinathan G, Zhang X, Luan X, Diekwisch TGH. Changes in Hox Gene Chromatin Organization during Odontogenic Lineage Specification. Genes. 2023; 14(1):198. https://doi.org/10.3390/genes14010198
Chicago/Turabian StyleGopinathan, Gokul, Xinmin Zhang, Xianghong Luan, and Thomas G. H. Diekwisch. 2023. "Changes in Hox Gene Chromatin Organization during Odontogenic Lineage Specification" Genes 14, no. 1: 198. https://doi.org/10.3390/genes14010198